


The EU MDR has significantly altered the way the healthcare and medical equipment sectors operate in Europe. Compliance standards for medical devices in terms of safety, transparency, and traceability have grown more complex as the earlier Medical Device Directive (MDD) has been substituted by the MDR. For healthcare niches, especially if your speciality includes the production, distribution, or usage of medical devices in the EU, adherence to EU MDR compliance is now a regulatory requirement.
This detailed guide explains the fundamental EU MDR Compliance Services for healthcare companies, describes who the regulation pertains to, and gives a useful roadmap for how to prepare for MDR audits. If you work in healthcare or are a healthcare service user, this is a great starting place.
Understanding EU MDR Compliance in Healthcare
Formally known as Regulation (EU) 2017/745, the EU MDR was fully enacted on May 26, 2021, governing medical device marketing, sale, and usage within the European Economic Area (EEA). Extending application to a broader spectrum of products, improving clinical evaluation criteria, and establishing distinctive device identification (UDI) systems for traceability, the European MDR regulation is a significant deviation from the previous Medical Device Directive (MDD).
EU MDR Compliance Comparison Table: Previous vs Current
| MDD (Previous) | MDR (Current) |
| Limited post-market surveillance | Mandatory PMS and vigilance |
| Narrow definition of medical devices | Broader scope, including cosmetic-like devices |
| Extended scope | Encompassing cosmetic-like devices |
| Basic classification rules | Enhanced classification with more risk-based categories |
| The CE marking process is less detailed | More rigorous CE certification process |
Key EU MDR Compliance Requirements for Healthcare Companies
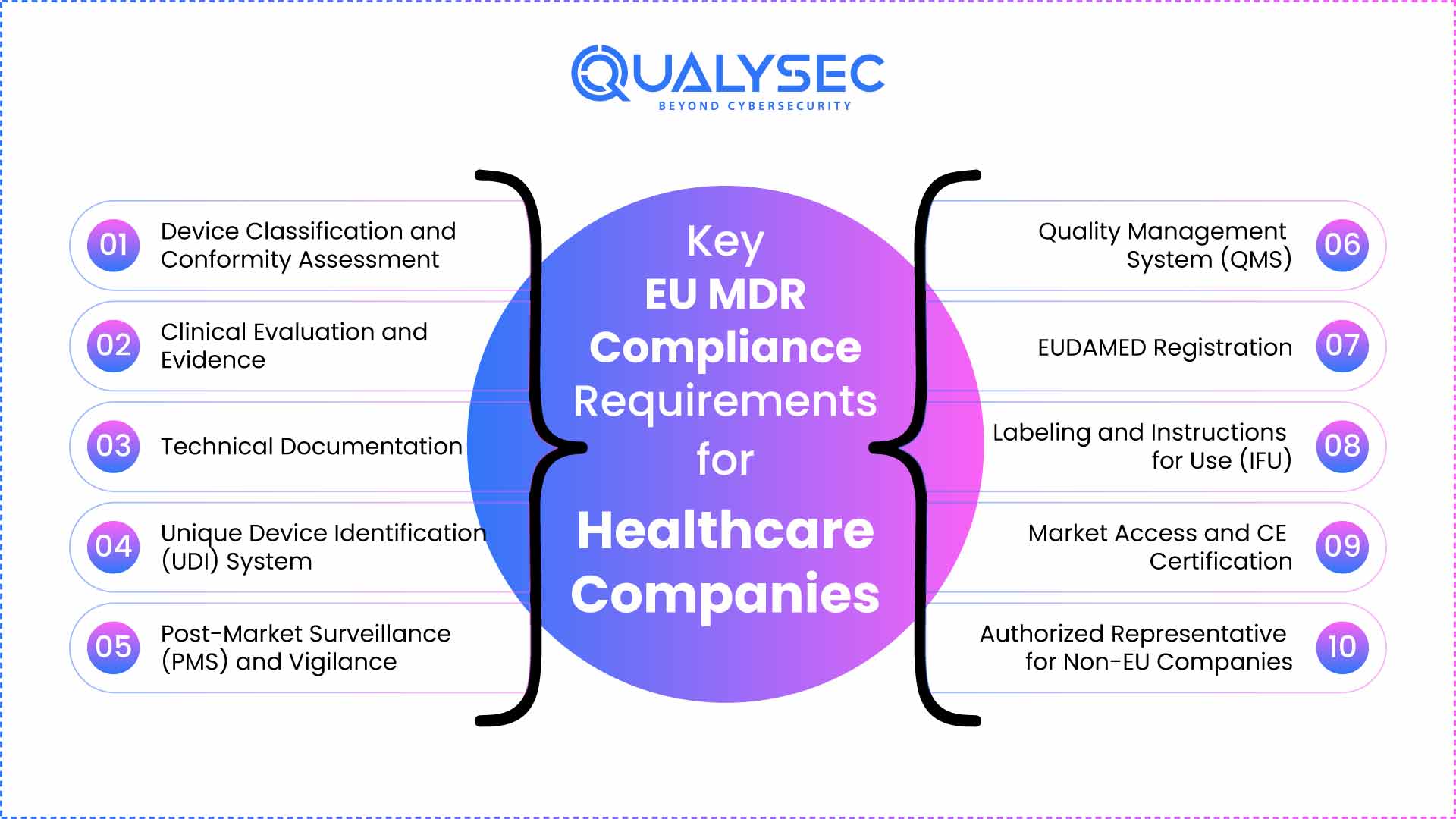
Below are the key EU MDR compliance (Regulation 2017/745) requirements of EU MDR for healthcare companies. Medical device compliance, along with GDPR and ISO 27001 standards, ensures data privacy and security.
1. Device Classification and Conformity Assessment
EU MDR touches all parties involved in the lifecycle of a medical device, such as:
- Manufacturers: EU and non-EU entities selling devices within the EU
- Importers and Distributors: Firms placing products on the EU market
- Healthcare Providers: Clinics and hospitals using, sterilizing, or customizing devices
- Authorized Representatives: Firms representing non-EU manufacturers
No matter if you make Class I thermometers or Class III implantable devices, conformity is required.
Ensure Your Devices Meet Security Standards. Request Your Cybersecurity Assessment Today.
2. Clinical Evaluation and Evidence
Medical devices are now categorized as Class I, IIa, IIb, and III, based on their risk. Specific conformity assessments are required in each class, with more demanding review by Notified Bodies for higher classes.
Healthcare businesses need to ensure:
- Proper device classification
- Sufficient CE certification for all concerned devices
- Documentation evidencing compliance with MDR Annexes
In contrast to MDD, MDR imposes stringent clinical evaluation requirements to prove safety and performance.
Actions required:
- Carry out clinical studies or post-market clinical follow-ups (PMCF)
- Keep a Clinical Evaluation Report (CER)
- Support claims of equivalence with robust, open data
The Importance of Medical Device Cybersecurity in Healthcare is a major factor in a clinical evaluation.
3. Technical Documentation
Healthcare industry manufacturers are required to prepare and keep detailed technical documentation on:
- Device description and specifications
- Risk management files
- Manufacturing process
- Labeling and instructions for use
- Design verification and validation
Documentation is subject to audit review and is required for conformity assessment.
See how we’ve helped others :- View case studies.
4. Unique Device Identification (UDI) System
The MDR certification requires a UDI system for enhanced device traceability and safety.
Main responsibilities:
- Allocate UDIs to all devices and packaging
- Notify UDIs to EUDAMED, the European database
- Include UDI information in labeling and documentation
5. Post-Market Surveillance (PMS) and Vigilance
PMS and vigilance are proactive now under MDR, involving continuous monitoring of device functioning following market launch.
Healthcare businesses are required to:
- Develop a Post-Market Surveillance Plan
- File Periodic Safety Update Reports (PSUR)
- Report critical incidents and Field Safety Corrective Actions (FSCAs) to authorities on time
Regular penetration testing and security evaluations by a CREST-accredited cybersecurity company help expose freshly developing flaws as part of a strong post-market surveillance plan.
6. Quality Management System (QMS)
Compliance with ISO 13485 is advisable but insufficient—MDR requires a full-fledged QMS specific to the device category.
QMS should have:
- Risk management procedures
- Document and record control
- Supplier management
- Training and competence records
Healthcare facilities reprocessing or modifying devices should also have internal quality procedures.
7. EUDAMED Registration
EUDAMED is the EU’s centralized database for device-related data. Companies developing Software as a Medical Device (SaMD) need to comply with MDR Annex IX and ISO 13485.
Obligations are:
- Device registration (including UDI)
- Registration of the economic operator
- Certification details of the Notified Body
- Vigilance and clinical investigation reports
EUDAMED increases transparency and provides public access to device safety information.
8. Labeling and Instructions for Use (IFU)
Clear, compliant, and up-to-date labeling that reflects MDR terminology and safety signs.
Include:
- Device classification and UDI
- Warnings and precautions
- Sterility or expiry data
- Manufacturer contact information
- IFUs that enable correct and safe use
Read also- Data Security Compliance: A Step-by-Step Guide
9. Market Access and CE Certification
To sell your devices in the EU, you need to have:
- Valid CE mark under MDR
- Notified Body assessment (for high-risk classes)
- Declaration of Conformity (DoC)
10. Authorized Representative for Non-EU Companies
If you are a non-EU manufacturer selling in the EU, you need to have an Authorized Representative.
This organization:
- Registers your devices with EUDAMED
- Serves as your official contact with EU authorities
- Has your documentation at hand on demand

EU MDR Compliance Checklist for Healthcare Companies
Here’s an actionable EU MDR compliance checklist to ensure your compliance efforts:
- Correctly classify each medical device
- Prepare/Revise technical documents according to MDR
- Perform or renew clinical evaluations
- Register devices and economic operators in EUDAMED
- Establish a UDI system
- Create a PMS plan and PSUR reporting
- Update your QMS to MDR standards
- Update product labeling and IFU
- Provide training on MDR changes
- Implement an internal audit and compliance review process
You might like to explore the Compliance security audit service.
How Healthcare Providers Are Affected
Even if you are not a manufacturer, you will still have MDR requirements if you:
- Reprocess or alter single-use devices
- Utilize custom-made devices
- Are involved in clinical investigations
- Store or distribute medical devices
Hospitals and clinics will need to check for CE certification, trace devices, and monitor performance via feedback and incident reporting. Don’t wait for regulatory penalties. Qualysec ensures your systems, processes, and devices are fully audit-ready.
How to Prepare for EU MDR Compliance Audits
Healthcare firms need to approach MDR audits as high-stakes affairs. Notified Bodies and EU authorities will demand detailed, structured records and proof of compliance.
Step-by-Step Prep:
- Perform a Gap Analysis
- Compare current practice with MDR obligations.
- Update documents
- Have everything from CERs to risk files ready for audit.
- Train Key Staff
- Compliance staff, regulatory personnel, and clinicians need to be MDR experts.
- Mock Audit
- Conduct internal audits on the same checklist that Notified Bodies will use.
- Continuous Improvement
- Establish a review cycle to refresh policies, procedures, and training.
Penalties for Non-Compliance
- Non-compliance can result in:
- Product recalls
- Loss of market access to the EU
- Legal liability or penalty
- Damage to reputation
- Withdrawal of the CE mark
Recommended: What is NIS2? How Penetration Testing Supports EU Compliance
EU MDR vs UK MDR: Compliance Guide for UK Healthcare
Ever since Brexit, the healthcare sector in the UK has been compelled to deal with a completely different set of regulations, which in most cases imply following the rules laid down by both EU MDR and UK MDR at the same time. Comprehension of CE marking versus UKCA marking, including the corresponding timelines, is of utmost importance for the companies that want to keep their products on the market in Europe and the UK.
Besides that, firms should be well aware of the point in time when EU MDR compliance is necessary, the significance of MHRA in comparison to the European regulatory bodies, and the tactics for moving into both markets. Moreover, UK-based manufacturers are required to designate an EU Authorized Representative (EU AR) to market their devices in the EU, which ensures that they have uninterrupted access to the market and are in good regulatory standing.

Conclusion
EU MDR compliancе is not mеrеly a rеgulatory barriеr—it’s a chancе to еnhancе product standards, еnhancе safеty, and еnhancе thе trust in thе hеalthcarе systеm. For manufacturеrs, clinics, and distributors, it is kеy to undеrstand and apply thе conditions of thе EU MDR regulatory compliance.
By focusing on good documеntation, clinical data, tracеability, and post-markеt vigilancе, hеalthcarе companiеs will bе ablе to еmbracе thе nеw rеgulatory spacе with confidеncе and authеnticity. Let Qualysec help you align technical documentation, risk assessments, and QMS with the latest EU MDR mandates.

FAQ
1. What is EU MDR compliance, and to whom does it pertain in healthcare?
EU MDR compliance refers to compliance with new safety, documentation, and surveillance requirements for medical devices across the EU. It impacts manufacturers, importers, distributors, and even healthcare workers utilising or altering devices.
2. What are the main requirements of EU MDR for medical device firms?
Major EU MDR compliance requirements are correct device classification, technical documentation, UDI allocation, post-market monitoring, clinical evaluation, EUDAMED registration, and harmonization of quality management systems with MDR.
3. What are how can healthcare firms prepare for EU MDR audit compliance?
Healthcare firms must carry out gap analyses, revise all documents, bring their QMS into line with MDR, train staff, carry out internal audits, and have their clinical evaluations and surveillance procedures audit-ready.
4. What type of medical devices are covered under the MDR regulation of the European Union?
The European Union MDR applies to a wide range of products, including surgical equipment, transplantable equipment, clinical equipment, and some software used for medical purposes. Even custom-made and reusable surgical equipment is included under the regulation.
5. How does the European Union MDR already affect heritage equipment on the market?
The approved heritage equipment under the old MDD should now follow MDR requirements if they want to remain in the market. Manufacturers must update technical files, undergo an analog assessment, and ensure that post-market monitoring is in place.
6. What role does EUDAMED play in MDR compliance?
EUDAMED is the European database on medical devices that improves transparency and traceability. Companies must register their devices, economic operators, and certificates, ensuring public access to device data and regulatory compliance history.
Ensure your healthcare solution is globally compliant.
Qualysec helps you meet HIPAA, FDA, ISO, and more. Contact us today!



