






As of early 2026, the FDA’s publicly updated list under its FDA Guidance on AI in Medical Devices shows 1,451 AI-enabled medical devices authorized for marketing in the United States since 1995, with approximately 295 new authorizations in 2025 alone. Radiology continues to dominate, accounting for about 76% of all authorizations, or roughly 1,104 devices.
The pace of change is starting to create pressure on the regulatory side, too. Review teams are dealing with software that does not always behave like traditional medical technology, while manufacturers are trying to ship products built on models that may continue evolving after deployment.
A conventional device is reviewed largely as a fixed product. AI systems are different. Performance can shift over time because of retraining, changing datasets, workflow differences, or model updates introduced after clearance. In some cases, the changes are intentional. In others, they are not obvious until the device is already in use.
FDA’s response has been to move toward a Total Product Life Cycle framework, where evaluation extends beyond the initial authorization itself. That approach now sits at the center of how the agency views AI-enabled medical devices and Software as a Medical Device (SaMD).
The sections below break down where that framework is heading in practice, including the January 2025 lifecycle draft guidance, the finalized PCCP guidance released later in 2025, and the submission issues that continue slowing reviews down.
What Does FDA Guidance on AI in Medical Devices Actually Cover?
Much of the current regulatory discussion specifically focuses on AI/ML-enabled medical devices and AI-enabled device software functions (AI-DSFs), which the FDA treats differently from general-purpose healthcare software.
Not everything labeled “AI guidance” from the FDA applies to medical devices. That distinction matters more than most teams realize, especially when teams are trying to determine which document actually governs a submission.
In January 2025, the FDA released two separate draft guidances. One came from CDER and focused on AI used to support regulatory decision-making for drugs and biologics. The other, which is more relevant here, came from CDRH and focused specifically on AI-enabled device software functions.
Under that draft guidance, an AI-DSF refers to a device software function using one or more AI models, where the model generates outputs, classifications, predictions, or recommendations from input data.
What the FDA is really moving toward is continuous oversight instead of one-time review. The expectation now is that safety and effectiveness hold up across the full operational life of the device, not only during the initial submission stage.
For AI systems, especially models capable of retraining or post-deployment updates, this creates a very different regulatory burden than older software products faced.
The FDA, Health Canada, and the UK’s MHRA originally published the 10 Guiding Principles for Good Machine Learning Practice (GMLP) in 2021 through an international regulator collaboration. FDA’s recent cybersecurity guidance documents continue to align closely with those principles, particularly around validation, data quality, lifecycle management, and multidisciplinary oversight.
| Track | Center | Focus | Key 2025 Document |
| AI in Medical Devices | CDRH | Device software functions | Draft Lifecycle Guidance, Jan 2025 |
| AI in Drug Development | CDER | Regulatory decision support | Draft Guidance, Drugs and Biologics |
Mitigate model risks and accelerate FDA compliance. Watch our healthcare AI security video now.
The Three FDA Premarket Pathways for AI Medical Devices
Most manufacturers already know that the three major FDA pathways exist. What becomes harder is figuring out how AI changes the expectations inside each one.
510(k) remains the dominant route by a wide margin. Roughly 96-97% of FDA-cleared AI medical devices have gone through this pathway historically. The process depends on demonstrating substantial equivalence to an existing predicate device, which becomes more difficult once the model introduces functionality that does not map neatly onto older products.
De Novo applies when the technology is novel but does not fall into a high-risk category. It represents a much smaller portion of AI authorizations, somewhere around 2-3%, but it matters because successful De Novo decisions can later become predicates for future FDA 510(k) submissions.
PMA sits at the highest end of the regulatory spectrum. These are Class III devices where clinical evidence requirements become significantly heavier. Most AI-enabled devices do not land here, but when they do, FDA expectations around validation, software documentation, and clinical performance are considerably stricter.
Pathway decisions usually come down to three variables more than anything else:
- Risk classification still drives the starting point. Most AI-enabled products fall into Class II, which generally points toward either 510(k) or De Novo.
- The existence, or absence, of a suitable predicate device often determines whether De Novo becomes necessary.
- Intended use frequently matters more than the underlying AI technology itself when the FDA makes classification decisions.
| Criteria | 510(k) | De Novo | PMA |
| Risk Level | Low to moderate | Low to moderate, novel | High, Class III |
| Predicate needed | Yes | No | No |
| Share of AI clearances | ~96-97% | ~2-3% | Under 1% |
| Clinical evidence | Often limited | Frequently required | Typically required |
Pro Tip: Selecting the wrong pathway early does not just delay timelines. In some cases, it forces teams to rebuild major sections of the submission package entirely.
FDA’s Total Product Lifecycle Approach and the January 2025 Draft Guidance
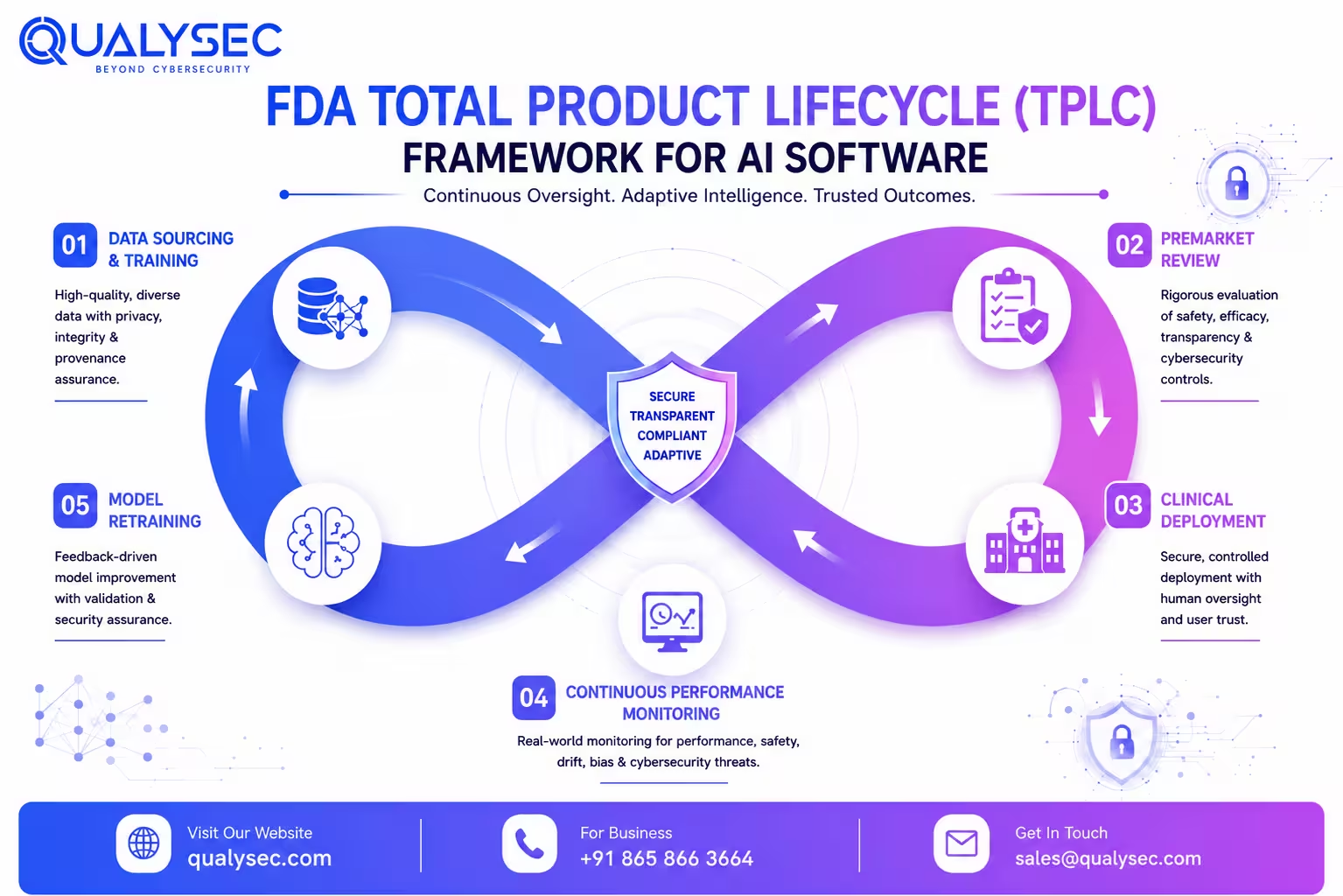
Issued on January 7, 2025, the draft guidance titled Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations marked one of the FDA’s clearest attempts to define how AI systems should be evaluated over time rather than only at clearance.
The central shift is a straightforward concept, even if implementation is harder in practice.
FDA no longer views authorization as the end of oversight. Retraining, performance drift, software updates, and changing real-world datasets all fall inside the broader Total Product Lifecycle framework.
What the guidance actually expects in a submission package:
- FDA expects manufacturers to explain what the model actually does, where it performs well, and where its limitations start becoming clinically relevant
- Training, tuning, and testing datasets need to be documented carefully. Reviewers increasingly want to understand how data was sourced, separated, and evaluated across the development process.
- Validation results are expected across demographic subgroups rather than only aggregate accuracy metrics. Age, ethnicity, sex, and race segmentation come up frequently during review now.
- Bias mitigation cannot stay theoretical. If known risks were identified during development, reviewers generally expect to see what was done about them and whether the mitigation changed outcomes in a measurable way.
- Human factors work matters more than many teams initially expect, especially in clinical environments where users may rely too heavily on AI-generated outputs under time pressure
- Labeling needs to explain system behavior in plain clinical terms, including situations where confidence, reliability, or output consistency may weaken
- The FDA is also putting more emphasis on postmarket monitoring plans capable of detecting performance drift outside controlled validation conditions
Importance of Early FDA Interaction
One thing regulatory teams still underestimate is the value of early interaction through the Q-Submission process. FDA feedback before a formal filing can prevent long review cycles later, especially for adaptive AI systems or products involving PCCPs.
Another distinction FDA continues evaluating closely is the difference between locked and adaptive models. Locked systems remain functionally static unless manually updated by the manufacturer. Adaptive systems introduce additional complexity because their behavior may evolve over time through retraining or continuous learning processes.
FDA scrutiny tends to increase further when continuous learning systems are designed to modify behavior after deployment using newly collected operational or clinical data.
Cybersecurity has also become tightly connected to lifecycle oversight expectations. FDA increasingly expects AI-enabled device manufacturers to address secure development, vulnerability management, and postmarket security monitoring as part of the broader governance picture rather than as isolated compliance tasks.
What Is a Predetermined Change Control Plan (PCCP) and Why Does It Matter for AI Devices?
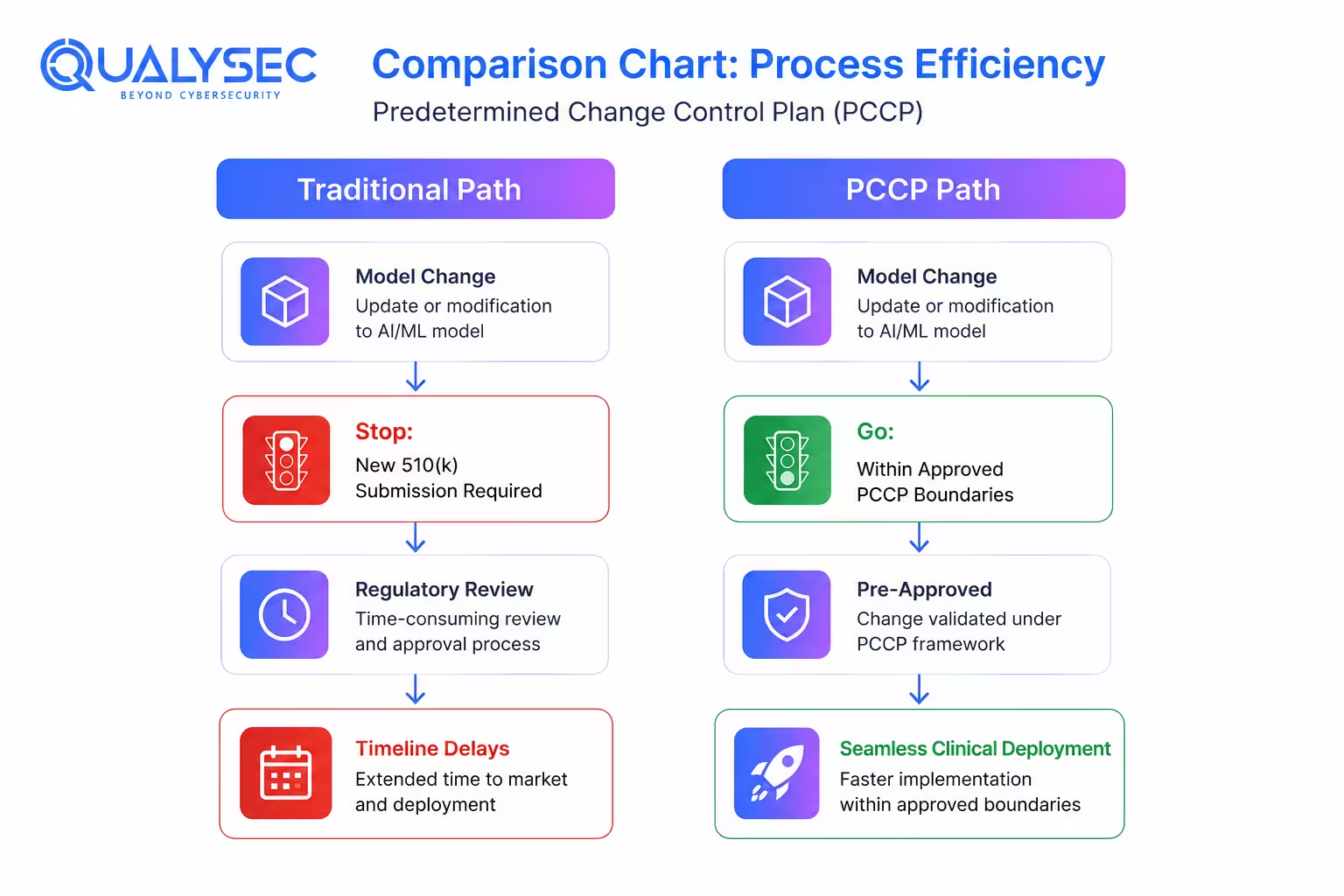
The August 2025 final PCCP guidance attempted to solve a problem that traditional device regulation was never designed for. AI systems evolve. Models get retrained, parameters change, and performance may improve or degrade after deployment.
Under older regulatory assumptions, many of those changes could trigger entirely new submissions.
A PCCP allows manufacturers to define certain anticipated modifications upfront during the original submission. If future changes stay within the authorized boundaries, an additional marketing submission may not be required.
PCCPs are still relatively new in practice, but most regulatory teams expect them to become far more common once adaptive AI systems move into wider clinical use.
Manufacturers now have a more practical framework for handling planned AI model changes after authorization. Under the FDA’s August 2025 final PCCP guidance, companies can include certain future modifications directly within the original 510(k), De Novo, or PMA submission instead of treating every adjustment as a separate regulatory event later on.
That only works if the planned changes stay inside the boundaries defined in the PCCP itself. Once modifications start affecting intended use, core functionality, or risk assumptions in ways that were not originally documented, the regulatory picture changes pretty quickly.
Core submission components usually include:
- A clear explanation of what kinds of model or software changes are expected over time, including limits on how far those modifications can go before they fall outside the original authorization scope
- Documentation showing how updates will actually be tested, reviewed, validated, and rolled out internally. FDA expects predefined performance checks here, not vague monitoring language.
- Safety impact analysis. That can include performance drift, labeling implications, clinician-facing transparency, cybersecurity considerations, and how post-deployment monitoring will be handled if the system behavior changes in real-world settings
FDA generally expects these plans to be specific and operationally realistic rather than broad conceptual descriptions.
FDA’s Growing Focus on Generative AI and Foundation Models
FDA’s AI oversight discussions are no longer limited to traditional machine learning systems. By late 2025 and into 2026, the agency had already started signaling increased attention toward foundation models and generative AI functionality integrated into medical devices.
Part of the problem is unpredictability. Traditional locked algorithms usually behave consistently when given the same inputs. Generative systems do not always work that way. Outputs can vary depending on prompts, context windows, retrieval layers, or changes introduced during fine-tuning.
That creates a different set of compliance problems compared to traditional ML systems:
- reproducibility of outputs across clinical settings
- hallucinated or fabricated responses
- transparency into how the generated outputs are formed
- clinician’s overreliance on systems that appear highly confident
- monitoring whether performance changes over time after deployment
The FDA still has not issued a standalone final guidance focused entirely on generative AI medical devices. Even so, companies working on LLM-based clinical tools are already seeing review conversations expand beyond traditional software validation questions.
A lot of the concern now sits around reliability in real clinical environments. Review teams are paying closer attention to output consistency, hallucination risks, cybersecurity exposure, traceability of generated responses, and whether clinicians could over-rely on recommendations that appear authoritative but are not always reproducible.
Internally, many regulatory and quality teams have started preparing submissions as if future generative AI oversight will become more demanding than what earlier AI-enabled software products faced.
Key Challenges in FDA Review and Compliance for AI Medical Devices
On paper, most regulatory teams already know what the FDA expects. The problem usually appears later, once those expectations have to be translated into a submission package that survives detailed review.
1. Bias and Dataset Representation
Bias tends to become one of the harder parts of review, partly because many datasets were never built with broad representation in mind to begin with.
FDA reviewers increasingly ask for subgroup-level validation instead of a single headline accuracy number. Once those breakdowns appear, weaknesses become harder to hide. Performance gaps across demographic groups are not automatically disqualifying, but reviewers usually want to see whether the issue was identified early and whether any mitigation work was attempted.
A lot of submissions struggle here because the underlying data simply was not designed for that level of analysis.
2. Transparency and Documentation Gaps
Transparency creates a different issue.
Some manufacturers provide detailed explanations covering validation methods, limitations, and model behavior. Others stay closer to high-level summaries. When documentation becomes thin, FDA review cycles usually become longer because reviewers start requesting additional clarification. In practice, that often translates into additional information requests, extended review holds, or multiple review cycle iterations before clearance.
3. Post-Deployment Monitoring Challenges
Performance monitoring after deployment remains another weak spot.
Validation results inside controlled environments only tell part of the story. Clinical workflows shift, patient populations change, and even infrastructure differences can influence model behavior over time. FDA’s lifecycle approach is increasingly built around the assumption that ongoing monitoring is necessary rather than optional.
4. Operational Weak Spots During Review
Review teams also tend to focus heavily on these:
- Cybersecurity concerns increasingly extend beyond patient data protection. FDA reviewers are paying more attention to adversarial attacks, compromised training pipelines and model manipulation risks.
- Documentation complexity grows quickly during AI submissions because datasets, validation outputs, software versions and clinical claims all need to stay connected and traceable.
- Intended use boundaries can drift over time, especially once products begin evolving after deployment.
- Traceability problems continue showing up more often than they should. Weak links between training data, validation evidence and final clinical claims raise questions that become difficult to resolve late in review.
Most of these issues do not automatically sink a submission. What they usually do is stretch timelines and increase the amount of review back-and-forth.
Don’t let regulatory holds delay your product launch. Schedule a compliance readiness review with a Qualysec expert.
Consult with our cybersecurity experts
Discuss your unique security requirements and discover how we can help your business.
How Medical Device Companies Can Navigate FDA AI Guidance and Build Compliance
There is no single checklist that gets you through this. But there is a logical sequence that separates companies that move efficiently through the process from those that keep cycling back.
I. Start With Risk Classification Early
Start with risk classification early. Where your device lands, Class I, II, or III, determines everything downstream. Pathway, evidence requirements, and postmarket obligations. Getting this wrong late is expensive.
II. Engage the FDA before the formal submission
The Q-Submission program exists precisely for this. For novel AI features, adaptive models, or anything involving a PCCP, an early pre-sub meeting can surface expectations that are not always explicit in the guidance documents. Manufacturers who skip this step often learn what the FDA wanted after they have already built the submission package.
III. Build Compliance Into the Design Stage
A few things that need to be built in from the design stage, not added later:
- GMLP alignment: multidisciplinary teams, representative training data, documented validation, transparency in model behavior. The 10 IMDRF principles are a practical framework here
- Bias evaluation and subgroup testing, done early enough to actually influence the dataset, not just reported after the fact
- Human factors studies, especially if clinicians will be acting on AI outputs in time-pressured environments
- PCCP planning, if the model is expected to evolve post-clearance
- Postmarket surveillance infrastructure that can realistically detect performance drift
IV. Adapting to the FDA’s Updated Quality Management System Regulation
Another shift companies are adjusting to is the FDA’s Quality Management System Regulation, which took effect in February 2026. The rule aligns U.S. quality requirements more closely with ISO 13485:2016. For AI-enabled devices, that has practical consequences. Risk management, software controls, complaint handling, validation records, and lifecycle monitoring are now expected to operate within a more connected quality framework rather than as isolated compliance activities.
V. Cybersecurity Expectations for AI-Enabled Devices
Cybersecurity deserves its own focus. AI-enabled devices carry specific risks, model integrity attacks, data poisoning, and vulnerabilities that traditional device security frameworks were not designed to catch. FDA reviewers increasingly expect AI-enabled device manufacturers to demonstrate secure development practices, vulnerability management procedures, and postmarket cybersecurity monitoring alongside core AI validation evidence.
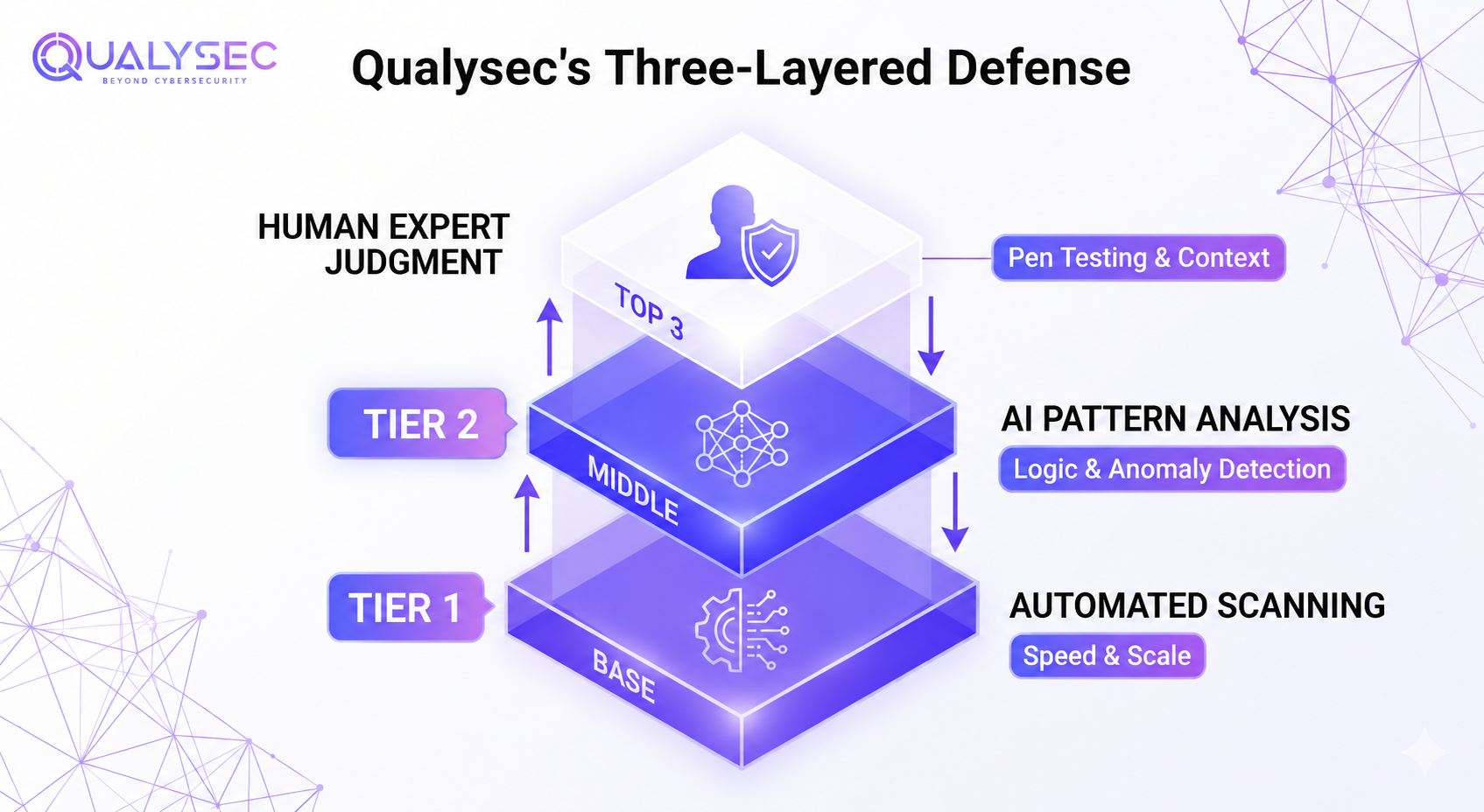
VI. Qualysec’s Approach to AI Device Security
Qualysec operates as a human-led, AI-powered cybersecurity company built around a three-layered defence system. Automated tools handle scale and speed at Layer 1. AI analysis catches complex patterns at Layer 2. Human experts bring judgment and creative thinking at Layer 3 that machines consistently miss. For medical device manufacturers, that combination of speed, depth, and human intuition matters, particularly when patient data and model integrity are both on the line.
Final Thoughts
FDA’s framework for AI medical devices is still evolving. The January 2025 draft guidance remains under review as of April 2026. But the direction is clear. Lifecycle thinking, bias accountability, adaptive change management through PCCPs, these are not future expectations. They are current ones.
The manufacturers moving fastest through review are usually not the ones with the most advanced models. They are the ones with stronger validation discipline, cleaner documentation, clearer intended use boundaries, and better lifecycle monitoring strategies. Cybersecurity is part of that picture too, not an afterthought.
Ready to navigate your FDA compliance roadmap with confidence? Book your free cybersecurity consultation today!
Secure your FDA AI compliance

Frequently Asked Questions
1. What is FDA guidance on AI in medical devices?
Two documents sit at the center of this right now. The January 2025 draft guidance covers lifecycle management for AI-enabled device software functions. The August 2025 final guidance deals specifically with PCCPs. Between them, they lay out what the FDA expects from the initial submission through everything that comes after deployment.
2. Which premarket pathway applies to AI devices?
510(k) is where most land, around 96-97% of clearances, historically. De Novo comes into play when there is no suitable predicate, and the device is genuinely novel. PMA is the highest bar, reserved for Class III devices, and clinical evidence is not optional there.
3. What is a PCCP, and when is it needed?
It lets manufacturers define upfront, at submission, what kinds of AI model changes they anticipate making post-clearance. If those changes stay within what was authorized, a new submission is not required. Particularly relevant when the model is designed to retrain or update after deployment.
4. What does the FDA look for in an AI device submission?
Model architecture and limitations, how training and testing data were sourced and split, subgroup performance validation, bias analysis, human factors data, labeling that reflects what the AI actually does, and a postmarket monitoring plan with some real teeth to it.
5. What are the biggest compliance gaps manufacturers face?
Biased or unrepresentative training data, validation reporting that lacks depth, postmarket surveillance that was never properly built out, and cybersecurity risks that AI systems carry, which traditional device frameworks were not designed to handle.














































































































































































































































































































































































































































































































































































































0 Comments