






Key Takeaways –
- EU MDR software development requirements include guidelines that every medical device software needs to follow to achieve CE marking and be sold in Europe.
- From software design to testing, release, and post-launch monitoring, these standards include cybersecurity in every phase.
- Certain standards, such as IEC 62304, IEC 62443, and ISO 14971, serve as the backbone of a compliant medical software development process.
- Modern-day hackers look for ways to exploit healthcare systems and steal data, making medical industry compliance necessary.
- The secure medical device lifecycle EU needs to follow secure coding practices, threat modelling, and Software Bill of Materials (SBOM) management continuously.
Introduction:
Imagine a scenario where a connected insulin pump, used by thousands of patients, contains a serious vulnerability. It will give hackers or attackers a free hand in terms of remotely increasing the dosage. In such cases, the medical device manufacturer faces a regulatory investigation and damage to its reputation. This could lead to huge fines or penalties that your organization takes years to recover from. Such cases are actually happening, and that’s why there is a need for the EU Medical Device Regulation to remain safeguarded from any potential attacks. This is where a Secure Medical Device Development Lifecycle under EU MDR becomes essential, helping manufacturers build security into every stage and minimize risks.
According to Palo Alto Networks’ Unit 42 IoT Threat Report, over half of connected medical devices were found to run on outdated software, exposing them to unpatched vulnerabilities.
The secure medical device lifecycle EU guidelines became fully applicable from May 26, 2021, and highlight the comprehensive measures to govern or regulate the medical devices and software. However, many medical software manufacturers fail to understand the importance of these requirements. As per EU MDR compliance requirements, the cybersecurity practices are no longer recommended practice in Europe, but legal requirements.
In case your medical device runs on software, the entire development and launch process needs to be secure from the very first lines of code. In this blog, we’ll understand the importance of security factors in software development as per the EU MDR standards.
What Is a Secure Medical Device Development Lifecycle?
A Secure Software Development Lifecycle (SSDLC) is a defined approach that integrates security into every stage of the medical software development cycle. In traditional medical devices, the security aspects used to be implemented in the very end phase, i.e., just before software deployment. On the other hand, as per EU MDR regulations, healthcare device secure coding practices need to have security (testing or vulnerability scanning) throughout the process to avoid any kind of loopholes for exploiters.
A secure medical device development lifecycle means that the requirements are predefined and introduced during the software development planning stage. This will assist in identifying the threats before the architecture is finalized with security practices in mind. Some of the testing parameters under secure medical devices include security validation and monitoring even after the product is available in the market.
General SDLC vs Secure Medical Device Lifecycle (EU MDR)
| Phase | Traditional SDLC | Secure Medical Device Lifecycle |
| Requirements | Functional requirements only | Functional + security + regulatory requirements |
| Design | Architecture and UX focus | Threat modelling + risk assessment (ISO 14971) |
| Development | Build features and functionality | Secure coding + static analysis (IEC 62304) |
| Testing | QA and user acceptance testing | Penetration testing + vulnerability assessment |
| Release | Product launch | Conformity assessment + CE marking + technical file |
| Post-Market | Bug fixes and updates | Continuous monitoring + patch management + PSUR |
What Are The Phases of a Secure Medical Device Development Lifecycle?
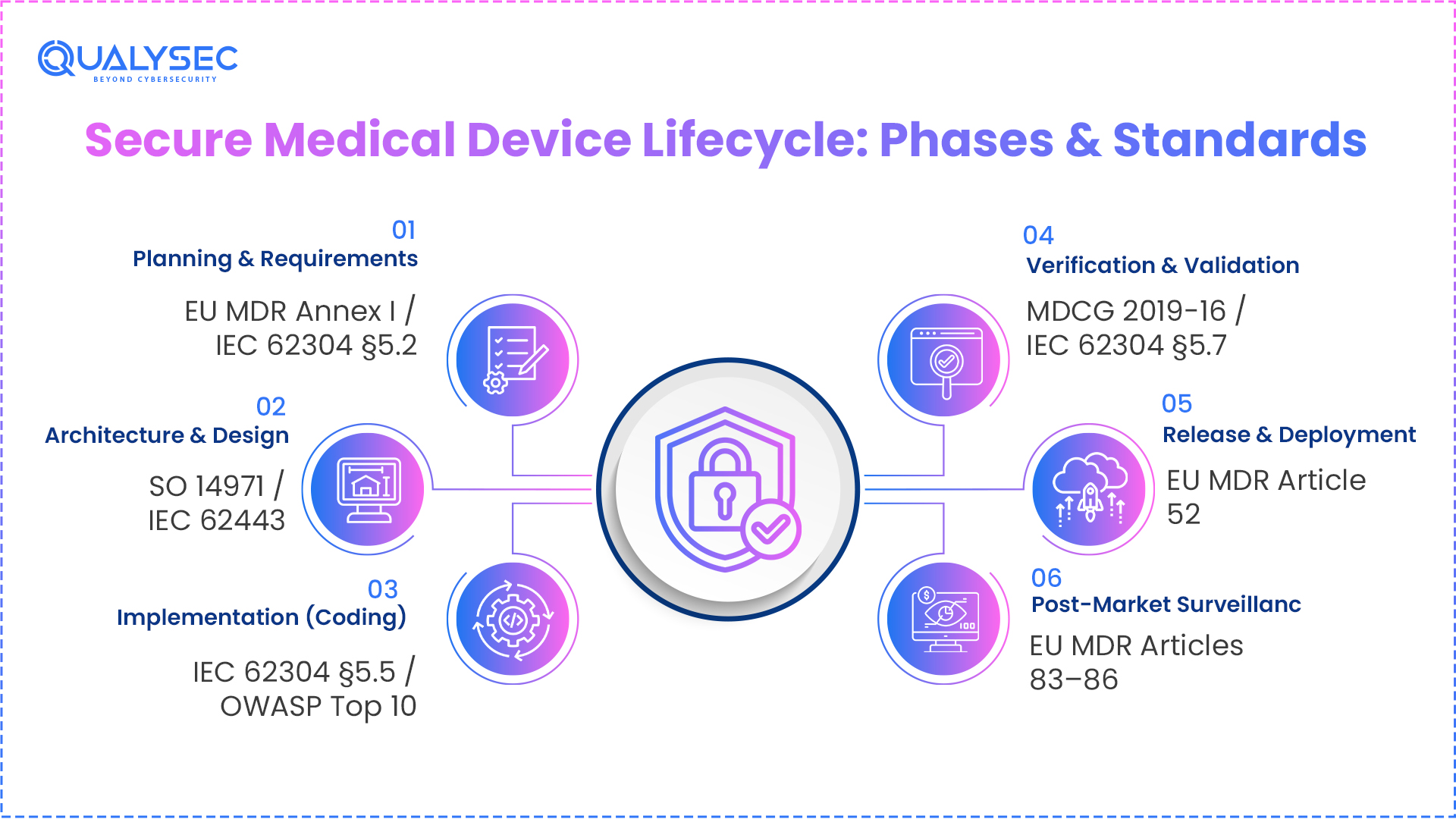
The ENISA Threat Landscape 2022 report identified healthcare as one of the most targeted sectors in Europe, with ransomware and data theft making up the majority of incidents affecting health organisations. This clearly highlights the importance of having secure development stages for medical devices and software in the EU.
A secure medical device development lifecycle is a continuous process that is associated with the entire product design, development, testing, deployment, and launch. Each phase of the software development lifecycle mandates that teams perform and document specific security activities as per the regulatory bodies.
Secure Medical Device Lifecycle Phases Mapped to EU MDR and Standards
| Phase | Key Security Activities | EU MDR / Standard Reference |
| Planning & Requirements | Define security requirements, identify regulatory scope, and classify software | EU MDR Annex I, IEC 62304 §5.2 |
| Architecture & Design | Threat modelling, security architecture review, data flow analysis | ISO 14971, IEC 62443 |
| Implementation | Secure coding, peer code review, and static analysis tools | IEC 62304 §5.5, OWASP Top 10 |
| Verification & Validation | Penetration testing, dynamic analysis, vulnerability assessment | MDCG 2019-16, IEC 62304 §5.7 |
| Release & Deployment | Conformity assessment, technical file compilation, CE marking | EU MDR Article 52 |
| Post-Market Surveillance | Continuous vulnerability monitoring, PSUR submission, and patch management | EU MDR Articles 83–86 |
What Are The Key Standards That Support Secure Development for Medical Devices?
Compliance with the secure medical device lifecycle EU doesn’t mean adopting any new practices. This means that the medical device manufacturers need to follow the international standards that regulatory bodies require. These standards do not replace EU MDR compliance; rather, they assist in achieving the same goal.
Key Standards for Secure Medical Device Development
| Standard | What It Covers | Relevance to EU MDR |
| IEC 62304 | Software lifecycle processes for medical devices | Directly referenced in the EU MDR technical documentation requirements |
| IEC 62443 | Cybersecurity for industrial and networked systems | Applies to connected and IoT-enabled medical devices |
| ISO 14971 | Risk management for medical devices | Mandatory for all CE-marked devices; covers cybersecurity risks |
| MDCG 2019-16 | EU guidance on software qualification and classification | Clarifies SaMD qualification, classification, clinical evaluation requirements, and cybersecurity documentation expectations |
| OWASP Top 10 | Common software vulnerabilities and mitigations | Widely used baseline for secure coding audits and penetration testing |
See How Leading Medical Device Companies Achieve Compliance. Explore real-world case studies on implementing secure development standards.

What Are Common Mistakes Manufacturers Make And How to Fix Them?
According to Cynerio’s 2022 State of IoMT Security report, around 1 in 4 medical devices (like imaging or surgical equipment) possess at least one vulnerability.
A lot of medical device/software manufacturers presume that they are non-compliant with the medical device secure development lifecycle because of missing security practices. The major problem lies in the disconnected security frameworks that later give loopholes to potential hackers and exploiters. In simple words, they treat cybersecurity or vulnerability testing as a one-time thing, instead of an ongoing practice.
Common Compliance Gaps and Best Practice Fixes
| Common Mistake | Why It Is a Problem | Best Practice Fix |
| No threat model at the design stage | Security vulnerabilities get baked into the architecture | Conduct STRIDE or PASTA threat modelling before coding begins |
| SBOM not maintained | Cannot track which components are affected by new CVEs | Automate SBOM generation using SPDX or CycloneDX formats |
| Security testing only at the end | Late-stage fixes are expensive and can delay launch | Shift-left i.e. integrate static analysis and security reviews throughout |
| Post-market monitoring ignored | Non-compliant with EU MDR Articles 83–86 | Set up continuous vulnerability monitoring and a defined patch policy |
| No patch management policy | Known vulnerabilities remain unresolved for months | Define patch cadence and document it in your technical file |
| Cybersecurity risk is not in the ISO 14971 assessment | Regulatory bodies will reject the technical file | Include cyber threats explicitly in risk management documentation |
How Does QualySec Help Medical Device Manufacturers Meet EU MDR Security Requirements?
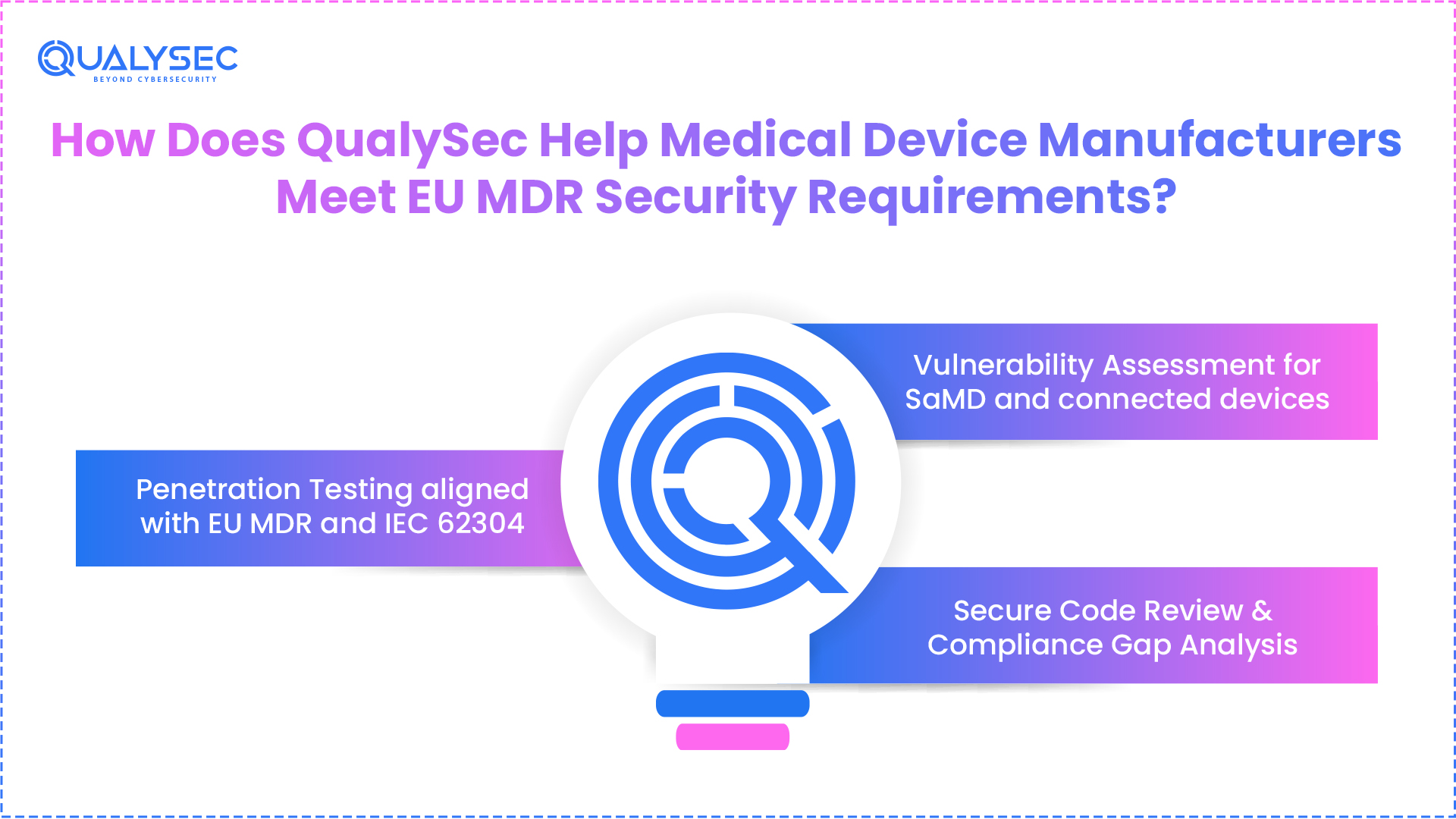
In 2026, the compliance of medical device or software manufacturers demands more than just basic intentions. The EU MDR software development requirements require a defined process of software testing, documentation, and continuous monitoring for security practices. While these seem to be easier said than done, the majority of in-house developers or testers cannot deliver the compliance.
Qualysec, the best cybersecurity company, specializes in assisting medical companies in the EU to achieve compliance as per the regulatory guidelines. This includes medical device manufacturers to create and maintain secure software/devices to stand up against the regulatory scrutiny. Here is what Qualysec Technologies provides the medical manufacturers operating under EU MDR:
a) Penetration Testing aligned with EU MDR and IEC 62304
At Qualysec, we provide human-led AI penetration testing services to find vulnerabilities and loopholes. Our team combines the best of human security intelligence with AI agents and automated scanners to find security issues. In simple words, our team will perform real-world-like attack scenarios against your medical device software, APIs, and connected infrastructure. Further, they document the findings of the results in the format as per the EU MDR guidelines.
b) Vulnerability Assessment for SaMD and connected devices
The Qualysec team identifies the known vulnerabilities across your medical device and software systems. Further, these vulnerability assessments include third-party libraries and align them according to the prioritization based on the patient risk impact.
c) Secure Code Review & Compliance Gap Analysis
Our engineering and testing team checked the source code against OWASP Top 10 and IEC 62304 requirements to identify the potential weaknesses. This will help in figuring out the issues even before they reach the testing phase of medical software development. Next, we will map the current development process against the EU MDR, IEC 62443, ISO 14971, and IEC 62304 standards.
If you are preparing for a conformity assessment, planning a new SaMD product, or responding to regulatory feedback on your technical file, QualySec can help you move faster and with more confidence.
Contact QualySec experts to review your current development lifecycle against EU MDR requirements.
Conclusion
Hence, the EU MDR software development requirements have changed the definition of security practices for medical device manufacturers in Europe. In reality, the medical device software manufacturer needs to take security compliance against set guidelines more seriously than ever. Failure to comply with the EU regulations on compliance to secure the medical device lifecycle will lead to fines and penalties.
As per the framework, IEC 62304 covers the management of the software lifecycle, ISO 14971 highlights the cybersecurity risk management, and IEC 62443 provides the cybersecurity framework for securing connected and networked medical devices. Any medical device and software manufacturer that is looking forward to conformity assessment needs to build security frameworks into the software development process. Rather than treating it just as a formality, the manufacturing companies need to focus on documentation and post-market surveillance.
Schedule a meeting with our experts to secure your medical devices and ensure EU MDR compliance.














































































































































































































































































































































































































































































































































































































0 Comments