






Cybersecurity has moved far beyond being a recommended practice. It directly influences whether your medical device reaches the market at all. If you are preparing for approval, you are no longer just building a safe product; you are proving that it can withstand real-world cyber threats. This is where understanding FDA vs EU MDR cybersecurity requirements becomes essential, as both frameworks expect clear, structured evidence of how your device manages and mitigates evolving security risks.
At the same time, you face a growing challenge. A single device must meet two very different regulatory mindsets. In the United States, the FDA expects structured, submission-focused cybersecurity evidence. In Europe, EU MDR leans on broader principles, with deeper scrutiny during audits by notified bodies.
This gap creates real risk. A misunderstanding can delay approvals, regulatory objections, or even refusal to accept your submission. With over 1.2 million connected healthcare systems exposed online as of August 2025, regulators are taking no chances.
In the sections ahead, you will see where these frameworks align and where they sharply differ.
Key Takeaways
- FDA cybersecurity compliance is required at the premarket stage with clearly defined deliverables.
- EU MDR embeds cybersecurity within Annex I and evaluates it during audits.
- The FDA strongly expects an SBOM as part of cybersecurity documentation in most submissions, while the EU MDR addresses it indirectly through documentation.
- FDA outlines clear expectations for patching and vulnerability handling.
- EU MDR relies on “state of the art” principles, with additional clarity provided through MDCG guidance documents.
- A practical approach is to build to FDA-level detail and align the same work with the EU MDR.
What Changed Recently in FDA vs EU MDR Cybersecurity
Things have shifted, and the shift becomes clear during documentation preparation. Regulators are no longer satisfied with surface-level security claims. They want clear proof, and they want it presented in a way that fits their review style.
In the US, the expectation leans toward structured evidence that can be checked quickly during submission. In Europe, the pressure shows up during audits, where your justification and consistency across documents are examined closely. If you treat both the same way and think about both the FDA vs EU MDR cybersecurity requirements are the same, gaps start to show.
FDA 2026 Guidance and QMSR Impact
The FDA has made cybersecurity part of your quality system, not a side activity. As part of FDA compliance, it requires you to handle security within your existing processes from day one, often implemented alongside standards like ISO 13485 to ensure consistency across development and risk management.
The introduction of the Secure Product Development Framework changes how you build your device. You are expected to consider security early, document your decisions, and demonstrate how each risk is addressed through design and testing. Reviewers look for that connection. If it is missing, it raises questions.
There is also more attention on how you manage risks over time. It is not enough to describe what you did before submission. You need to show how you will keep tracking and addressing issues as the device is used.
One detail that catches many teams off guard is the initial screening step. If your cybersecurity documentation is incomplete or unclear, your file may not move forward at all.
EU MDR Cybersecurity Interpretation via MDCG
On the European side, the legal framework has not changed much, but expectations have become clearer through MDCG 2019 16 Rev.1. This guidance gives you a better sense of what reviewers look for when they assess cybersecurity within your technical documentation. It makes EU MDR compliance more structured and easier to align with during the approval process.
Notified Bodies now look for concrete proof of how your device handles security risks. This includes penetration testing results, secure patch management, including validation of update integrity, and controlled deployment mechanisms. These details help demonstrate that your security measures work in practice.
Each Notified Body applies its own interpretation during audits, so your documentation needs to tell a complete and consistent story to avoid delays.
Regulatory Scope Difference: FDA Cyber Device vs EU MDR General Cybersecurity Requirements

One of the first differences you will notice lies in how each region defines the scope of cybersecurity. The FDA clearly identifies a category called “cyber devices,” while the EU MDR applies broader requirements across all devices without isolating a specific category in the same way.
FDA’s Defined Cyber Device Scope
The FDA draws a clear line around devices that fall under cybersecurity scrutiny. Devices that include software, have network connectivity, and present cybersecurity risks are typically classified as “cyber devices” under FDA guidance. Once classified, you are expected to provide dedicated cybersecurity evidence as part of your submission.
This is not treated as optional or supportive information. Reviewers look for structured proof that security risks have been identified and tested. If that evidence is missing or incomplete, your submission can be rejected.
EU MDR’s Broad and Implicit Cybersecurity Scope
EU MDR does not label devices under a separate cybersecurity group. You still need to cover security, but it sits within different parts of your documentation rather than one defined section.
You will deal with it through your software lifecycle work, your risk management file, and the controls you use to protect data and system behaviour. Reviewers expect these pieces to make sense together when they read your file.
When the audit happens, the Notified Body goes through your documents line by line. They look at how you handled risks, why certain decisions were made, and whether everything connects properly. Since there is no fixed format, how you explain your approach can either smooth things out or slow the process down.
Cover FDA and EU MDR requirements with complete medical device testing and compliance support. Contact us today.
Achieve compliance faster with expert guidance
See how expert guidance helps you achieve compliance faster.
FDA vs EU MDR Cybersecurity Requirements – Side-by-Side Breakdown
Risk Management and Threat Modelling Differences
A closer look at both approaches shows how expectations diverge:
| Aspect | FDA Approach | EU MDR Approach |
| Risk Perspective | Treats cybersecurity threats as reasonably foreseeable risks that must be clearly identified | Requires risks to be managed in line with current state-of-the-art practices |
| Threat Modelling | Requires structured threat modelling with documented methodology | Does not explicitly mandate formal threat modelling, but equivalent risk analysis is expected as part of risk management. |
| Documentation Depth | Expects detailed, organised records showing how threats were identified and controlled | Looks for evidence within the overall risk management file without a fixed format |
| Evaluation Focus | Checks whether a defined method has been followed and documented properly | Focuses on whether risks are addressed effectively in the outcome |
In simple terms, the FDA expects you to show how you reached your conclusions step by step. EU MDR focuses more on whether your approach holds up when reviewed.
Premarket Submission vs Technical Documentation
The FDA cybersecurity guidance for medical devices lays out what needs to be included in a submission, with clear expectations around structure and evidence. Under EU MDR, the emphasis moves toward how your technical documentation supports safety and performance during review.
| Aspect | FDA Approach | EU MDR Approach |
| Submission Type | Premarket submission with dedicated cybersecurity sections | Technical documentation reviewed during conformity assessment |
| Required Evidence | Cybersecurity risk reports, architecture diagrams, testing evidence, and SBOM | Documentation aligned with Annex I and Annex II requirements |
| Structure | Defined documentation format and content | Flexible structure based on how the technical file is organised |
| Review Style | Checked during submission screening and formal review | Assessed through audits and technical documentation evaluation |
| Key Expectation | Complete cybersecurity evidence presented upfront | Strong alignment across documents during assessment |
SBOM Requirements and Software Transparency
| Aspect | FDA Approach | EU MDR Approach |
| SBOM Requirement | Mandatory as part of submission expectations | No direct requirement is defined in the regulation |
| Component Visibility | Must list all third-party and open source components | Expected to demonstrate control over components through documentation |
| Supply Chain Insight | Explicit disclosure of software dependencies is required | Addressed indirectly through supply chain and risk management records |
| Key Expectation | Clear and complete SBOM included in submission | Evidence of software transparency spread across the documentation |
With the FDA, you need to present a full breakdown of software components without gaps. Under EU MDR, reviewers look for how well you track and manage those components, even though a formal SBOM is not explicitly required.
Cybersecurity Testing Expectations
| Aspect | FDA Approach | EU MDR Approach |
| Testing Evidence | Expects comprehensive security testing, which may include penetration testing, vulnerability assessments, and other validation methods based on device risk. | MDCG guidance points toward fuzz testing, vulnerability scanning, and penetration testing |
| Reporting Style | Expects detailed test reports with clear methodology and results | Focuses on whether testing methods are appropriate and justified |
| Validation Approach | Structured reporting with traceable outcomes | Method-driven validation based on how risks are addressed |
| Key Expectation | Clear documentation that explains how testing was performed and what was found | Strong justification of testing choices and coverage during review |
This may also extend to firmware-level controls such as secure boot and integrity validation for embedded systems.
FDA reviewers look for clear proof of what you tested and what came out of it. Under EU MDR, your healthcare device security testing approach must be well-justified, risk-based, and consistently documented to show it aligns with the risks your device carries.
Postmarket Surveillance and Vulnerability Handling
| Aspect | FDA Approach | EU MDR Approach |
| Vulnerability Monitoring | Defined process to track and assess vulnerabilities | Covered within postmarket surveillance activities |
| Disclosure Process | Coordinated vulnerability disclosure handled through a formal process | Managed within general vigilance and reporting activities |
| Risk Updates | Security issues handled through dedicated cybersecurity workflows | Risk management is updated continuously through PMS |
| Clinical Impact | Not directly linked to clinical evaluation | Findings feed into clinical evaluation and ongoing safety assessment |
| Key Expectation | Separate cybersecurity processes after release | Security is handled within the overall lifecycle |
The FDA places clear emphasis on having dedicated processes for vulnerability tracking and disclosure once the device is in use. EU MDR connects these activities with ongoing postmarket work, where updates influence both risk management and clinical evaluation.
Strengthen your premarket and postmarket surveillance and vulnerability handling with expert healthcare device pentesting. Watch our video to see how it works.
https://www.youtube.com/watch?v=arXxp7MRZbI
Patch Management and Update Timelines
| Aspect | FDA Approach | EU MDR Approach |
| Timeline Requirement | Expects timely remediation based on risk severity, without prescribing fixed timelines. | No fixed timelines defined |
| Patch Handling | Time-bound actions based on the severity of vulnerabilities | Updates handled based on risk and impact |
| Compliance Style | Driven by clear timelines and deadlines | Guided by lifecycle-based risk decisions |
| Key Expectation | Timely fixes aligned with predefined categories | Updates applied when risk justifies action |
The FDA puts clear time limits on fixing vulnerabilities, with faster action required for critical issues. EU MDR leaves the timing open, so decisions depend on the level of risk and how it affects the device in real use.
Labelling and IFU Cybersecurity Requirements
| Aspect | FDA Approach | EU MDR Approach |
| Labeling Content | Cybersecurity details included as part of device labelling | Information provided within the IFU and supporting documents |
| Purpose | Communicates known risks and safe usage conditions | Defines how the device should be used in a secure environment |
| Regulatory Impact | Missing details can lead to misbranding concerns | Gaps can raise issues during conformity assessment |
| Key Expectation | Clear communication of security-related risks | Practical instructions to support secure operation |
In the FDA context, the focus leans toward informing users about risks and safe handling. Under EU MDR, the emphasis moves toward how the device should be used and maintained securely in real settings.
Documentation Differences That Impact Approval Timelines
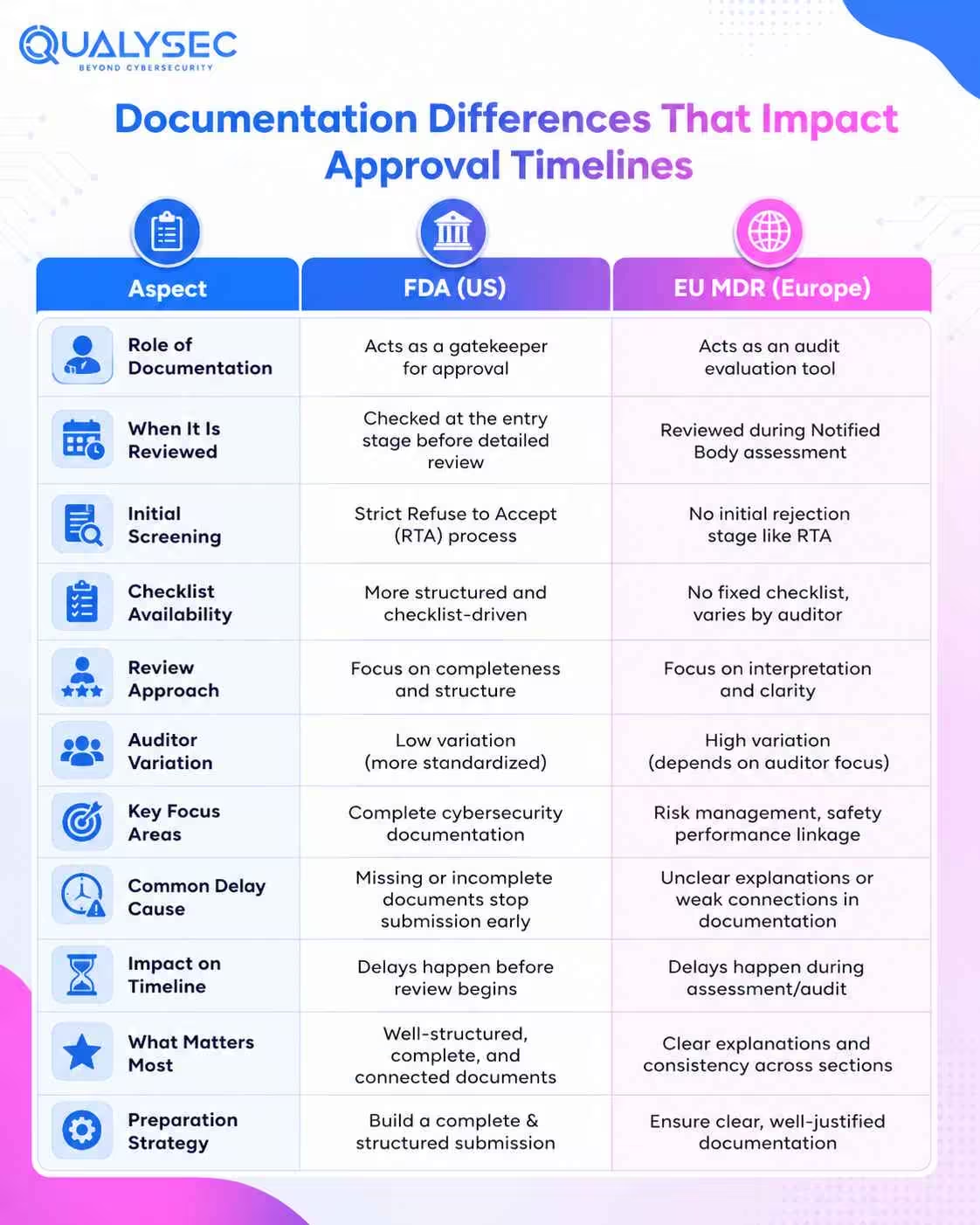
FDA Documentation as a Gatekeeper for Approval
For FDA submissions, cybersecurity documentation is checked right at the entry stage. It is not something that gets reviewed later. If required elements are missing, the submission can be stopped under Refuse to Accept before any detailed review begins.
This makes documentation quality critical. Everything needs to be complete, structured, and clearly connected. Even small gaps or unclear sections can delay progress before the evaluation actually starts.
EU MDR Documentation as an Audit Evaluation Tool
Under EU MDR, documentation is reviewed during the Notified Body assessment rather than at an initial screening stage. The way your file is read can influence the outcome more than you might expect.
There is no fixed checklist guiding the review. Different auditors may focus on different areas, which brings variation into the process. Some may go deep into risk management, and others may question how security links with performance and safety.
This becomes more evident when you look at global medical device cybersecurity regulations. Progress depends on how clearly your documentation explains decisions and connects across sections, not just what is included.
Practical Impact on Manufacturers
Delays come from different reasons in each region. In the US, missing or incomplete cybersecurity documents can stop your application before the review begins. In Europe, delays come from unclear explanations or gaps in how your documentation is understood during assessment.
You need to prepare in two ways. One is building a complete and well-structured submission. The other is writing clear explanations that are easy to follow during the audit.
Avoid delays caused by documentation gaps. Ensure your FDA 510(k) requirements and EU MDR technical file are aligned from the start. Check our sample pentest report.
Get a Free Sample Pentest Report

How FDA vs EU MDR Differences Shape Cybersecurity Strategy
Why the FDA Forces Structured Cybersecurity Programs
A clear difference appears in FDA vs MDR medical device regulations when you focus on how cybersecurity is handled. The FDA side pushes teams toward structured processes and clear documentation from the beginning.
SPDF brings security into the design stage itself. This includes practices such as secure coding, threat modelling, and architecture-level risk controls embedded throughout development. Teams need to think about risks early and test them properly. They also need to keep records that connect each step to the product. If something is missing or does not link clearly, it can create issues during review.
Why EU MDR Requires Flexible, Evidence-Based Security
EU MDR does not tell you exactly how to handle cybersecurity. That creates a different kind of pressure during review. Two teams can follow similar controls and still get very different outcomes. One clears the audit without much trouble. The other gets questioned in detail. The gap usually comes from how decisions are explained.
Reviewers look for clear reasoning. Why was a control chosen? How a risk was judged. Whether the level of protection actually fits the device. If that logic is not clear, it slows things down.
Building a Unified Approach for Both Regulations
Start with FDA-level documentation as your base, then map the same content to EU MDR Annex I requirements. Keep your risk management, postmarket activities, and technical documentation aligned so everything tells the same story.
Role of Penetration Testing in FDA vs EU MDR Compliance
FDA
- Requires structured penetration testing reports
- Reports must clearly define the scope
- Methodology needs to be explained
- Findings must be documented in detail
EU MDR
- Recognised as a state-of-the-art validation method under MDCG
- No fixed reporting format
- Evidence should support security claims and risk handling
The key difference lies in how each region looks at penetration testing. The FDA focuses on well-documented reports, where every detail is clearly presented. EU MDR, on the other hand, looks at how well the testing supports real-world risk handling. In practice, penetration testing helps cover both sides when it is planned properly and documented in a clear and meaningful way.
How Qualysec Supports FDA and EU MDR Cybersecurity Compliance
When you look at a medical device cybersecurity standards comparison, one thing becomes clear. Meeting both FDA and EU MDR expectations requires more than basic testing. It needs depth, clarity, and results that hold up during review.
Qualysec delivers penetration testing aligned with both regulatory approaches, covering IoT medical devices, APIs, cloud infrastructure, and embedded systems.
A key takeaway:
- Risk-based vulnerability reports
- Clear reproduction steps
- Practical remediation guidance
This supports:
- FDA submission requirements
- EU MDR technical documentation
A hybrid testing approach ensures:
- Low false positives
- Real-world attack simulation
Post assessment support includes retesting and consultation to align findings with compliance expectations.
Conclusion
Two regulators, two ways of looking at the same device. One looks for clear, structured proof before anything moves ahead. The other takes a closer look during review and questions how well everything holds together. This difference reflects FDA vs EU MDR cybersecurity requirements, where expectations vary in how evidence is presented and assessed. If your cybersecurity work is built with both in mind from the beginning, things move more smoothly. If not, gaps tend to surface when it matters most.
Don’t let cybersecurity gaps slow down your approval. Talk to Qualysec and get your device ready for both FDA and EU MDR review.
Talk to our Cybersecurity Expert to discuss your specific needs and how we can help your business.

FAQs
1. How do FDA vs EU MDR cybersecurity requirements differ?
The FDA follows a structured approach where specific cybersecurity evidence is reviewed during submission. EU MDR works differently, where security is assessed through broader documentation during audits. The difference comes down to how the evidence is presented and evaluated.
2. Does the FDA require SBOM, while MDR does not?
Yes. The FDA requires an SBOM as part of submission expectations, including all third-party and open source components. EU MDR does not explicitly ask for it, but similar details are expected through the supply chain and risk documentation.
3. Which regulation is stricter for medical device security?
Neither is stricter in a simple sense. The FDA is more rigid in documentation and structure, while the EU MDR can be challenging due to interpretation during audits.
4. How can manufacturers comply with both regulations?
A practical way is to build documentation to FDA-level detail, then align the same material with EU MDR requirements. Consistency across risk management, testing, and technical documentation helps reduce gaps.
5. What cybersecurity testing is required for both regions?
Both expect strong testing practices. This includes penetration testing, vulnerability assessments, and validation of security controls. The FDA focuses more on detailed reporting, while the EU MDR focuses on whether the testing supports the device’s risk profile.











































































































































































































































































































































































































































































































































































































0 Comments