






FDA 510k clearance is the most common regulatory route by which manufacturers of medical devices acquire legal authorization to market their products within the American market. Under this premarket notification process, companies must be able to prove that their new device is substantially equivalent to a predicate device that has already been legally sold in America. Most of the medical devices used in hospitals, clinics, and other health care facilities around the country are regulated by the FDA 510k clearance system.
The importance of understanding 510k medical device regulations cannot be underestimated for manufacturers seeking market access. This regulatory system covers more than 95 percent of the total number of medical devices sold in the United States, and 510k clearance knowledge is critical to succeeding in this industry. Unlike other approval pathways that involve extensive clinical testing, 510k clearance focuses on comparative analysis with existing devices. The 510 k process has provided manufacturers with simplified access to the market through a regulatory system. Overall, it serves as a guideline for discussing FDA 510 (k) process requirements, their challenges, and how to navigate complex regulatory issues satisfactorily.
What is FDA 510(k) Clearance and Why Does It Matter?
FDA 510k clearance is called a premarket notification. Further, it is a process followed under the Food, Drug, and Cosmetic Act at 510(k). This is achieved by requiring medical device manufacturers to report to the FDA at least 90 days before selling their products in the United States. The general aim of clearance 510k is to specify that a new device is substantially similar to a predicate device, which is already being marketed.
Approximately 95-98% of all medical equipment marketed in the US comes under the 510 k approval. This has made it the most significant regulatory route for medical device organizations. Unlike PMA approval, the FDA 510 (k) process does not require extensive clinical trials and safety data to demonstrate safety and efficacy, but shows a similarity to previously approved devices.
Request a compliance audit for your medical device!
Key Features of 510(k) Clearance:
- Substantial Equivalence: Your device must be similar to an already approved predicate device
- 90-Day Review Period: FDA has 90 days to review your submission
- Cost-Effective: Less expensive than the PMA approval process
- Faster Market Entry: Quicker pathway compared to clinical trials
- Class II Devices: Primarily for moderate-risk medical devices
- No Clinical Testing Required: In most cases, clinical data is not mandatory
The 510k medical device classification consists of 3 risk categories. Class I devices are low risk and usually do not require 510k clearance. Class II devices typically require 510k clearance, whereas Class III devices typically necessitate more rigorous PMA approval.
| Device Class | Risk Level | Approval Process | Examples |
| Class I | Low Risk | Exempt from 510(k) | Tongue depressors, bandages |
| Class II | Moderate Risk | 510(k) Clearance | Hip implants, pregnancy tests |
| Class III | High Risk | PMA Approval | Pacemakers, heart valves |
Must Read: FDA 510(k) Guidance: Step-by-Step Cybersecurity Compliance for Medical Devices.
How Does the 510(k) Submission Process Work?
The FDA 510 k process begins with a manufacturer identifying a predicate device with the same intended use as that of the new product. The manufacturer will then need to create extensive documentation demonstrating substantial equivalence. This documentation includes technical specifications, performance information, labeling information, and safety testing information.
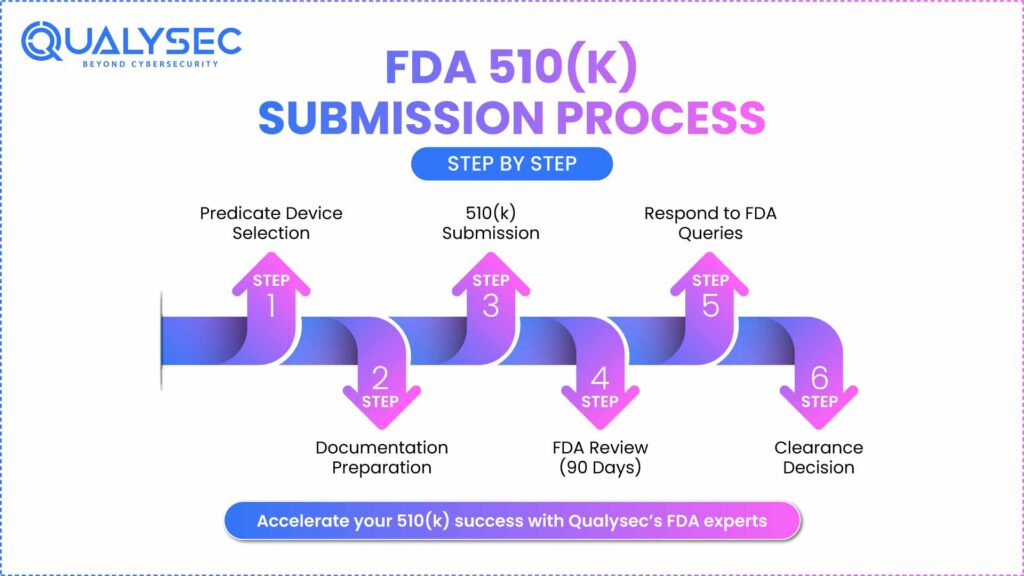
Step-by-Step 510(k) Process:
- Predicate Device Selection: Choose an appropriate comparison device already cleared by the FDA
- Documentation Preparation: Prepare all necessary technical and safety information.
- Submission Creation: Prepare the formal 510(k) notification document
- FDA Review: Agency reviews submission within 90 days
- Response to Questions: Address any FDA requests for additional information
- Clearance Decision: FDA issues clearance or denial
The 510 k approval process generally occurs within the time range of 90 to 150 days. Further, it depends on the complexity of the device and the quality of the submission. Manufacturers can accelerate this process by thoroughly documenting all FDA requirements before starting.
Essential Components of a 510(k) Submission:
- Device Description: Detailed technical specifications and intended use
- Predicate Device Comparison: Comprehensive analysis showing substantial equivalence
- Performance Testing: Bench testing, biocompatibility, and sterilization data
- Labeling: Proposed product labels and instructions for use
- Information on manufacturing: Quality control procedures and plant description.
- Risk Analysis: Evaluation of the possible risks and the remedies.
What Are the Major Problems with 510(k) Clearance?
Most people criticize the FDA 510 k process because it allows potentially dangerous devices into the market without any safety testing. Recent reports show that devices undergoing 510(k) clearance face 11.5 times more recalls than devices approved through the PMA process.
One of the biggest challenges facing many manufacturers, particularly those that develop innovative technologies, is finding suitable predicate devices. The 510k medical device pathway involves the identification of appropriate comparison devices that have the same intended use and are technologically close to each other. This may be especially challenging in the case of breakthrough technologies with no apparent market precedent.
Read more on FDA Cybersecurity Guidelines for Medical Devices 2025.
Major Challenges in the 510(k) Process:
- Limited Safety Testing: No clinical trials required for most 510 clearance submissions
- Predicate Device Dependencies: Must find a suitable comparison device for what is 510k clearance
- Recall Risk: Higher likelihood of post-market safety issues compared to PMA devices
- Innovation Constraints: May discourage breakthrough technologies in 510k medical device development
- Regulatory Uncertainty: Subjective substantial equivalence determinations in FDA 510k clearance
- Quality Control Issues: Insufficient oversight of manufacturing processes post-clearance
In 2011, the Institute of Medicine said that the 510 k approval process was fundamentally defective and that a different system must replace it. The report found that many moderate-risk devices were inflicting serious harm on patients. Consumer Reports revealed that, in many cases, manufacturers conduct safety testing on medical devices using the bodies of unaware patients.
Another major problem is the use of problematic predicate devices. Manufacturers can obtain devices with 510k clearance that are substantially similar to devices that were never shown to be safe and effective. Worse still, the same predicate devices can be exploited to legitimize new device approvals despite known design flaws or safety recalls of the device.
In most cases, the FDA 510 k process is also not transparent. The Journal of the American Medical Association published research that the FDA only publicly provides data used to establish substantial equivalence in 16 percent of devices cleared. This is where transparency fails, and so the medical practitioners and patients are unable to make decisions regarding the safety of the devices.
Need an FDA Agent? Learn about our FDA Agent Services here.
Why is Qualysec the Leading FDA 510(k) Clearance Partner in the USA?
In the complicated FDA 510k clearance process, having a team of skilled regulatory consultants may be the difference between an easy win and a delay that costs a lot. Qualysec is the leading consulting company in 510k clearance submissions and medical device regulatory compliance in the United States.
Qualysec has over ten years of experience in assisting medical device companies to navigate the FDA 510 (k) process successfully. The reason being, our group of former FDA reviewers and regulatory professionals knows precisely what the agency is seeking in 510k medical device submissions. Our clients have had a first-time approval rate of 98 percent, which is above the industry average.
Why Choose Qualysec for Your 510(k) Submission:
- Former FDA Reviewers: Our team includes ex-FDA professionals who understand agency expectations
- 98% Success Rate: Industry-leading first-time approval rate for 510k clearance submissions
- Comprehensive Services: End-to-end support from predicate device selection to clearance
- Fast Tracked Schedule: 110 days on average to approve a project, compared to the industry average of 150 days.
- Cost-Effective Solutions: Pricing is transparent, with no hidden charges for the 510k clearance guidance.
- Ongoing Support: Post-clearance compliance and quality system maintenance
We have a successful process that starts with an evaluation of your hardware and market environment. We assist in determining the most suitable predicate devices and in formulating a convincing, substantial equivalence argument that appeals to the FDA reviewers. The submissions prepared by our writers and regulatory professionals are clear, detailed, and in line with all the existing guidelines of the FDA regarding FDA 510k clearance.
The success stories of clients served by Qualysec span a wide range of medical devices, including both surgical and diagnostic equipment. We understand the specific challenges each device type presents and tailor our approach accordingly. We have the advantage of having a large database of successful 510 (k) approvals and an excellent knowledge of what the FDA reviewer likes.
The 510k medical device approval continues to change fast. FDA constantly updates its guidance documents, creates new requirements, and alters review processes. Qualysec stays above these shifts, keeping our clients informed on all the latest regulatory intelligence and strategy.
Ready to streamline your 510(k) submission process? Contact Qualysec today for a free consultation and discover how our expertise can accelerate your path to market.


































































































































































































































































































































































































































































































































































































































































































0 Comments