






FDA says 510(k)s submitted after October 1, 2023, must be submitted electronically using eSTAR, unless exempted. This move has led to a consistent approach to data collection during the initial acceptance period, but added to the technical review burden in the substantive decision-making phase. Moreover, recent financial reports show the average Total Time to Decision (TTD) for non-automated traditional FDA 510k timeline has now settled out at 140-170 calendar days, suggesting that in the current environment, the key to bypassing the “Refuse to Accept” (RTA) backlog that we saw in previous fiscal years is by providing quality technical documentation.
In this new environment, to obtain a “Substantially Equivalent” (SE) decision, manufacturers are recommended to embrace a more disciplined approach to their premarket notifications, one that is focused on data and quality.
Step-by-Step Process
| Phase | FDA Goal (Calendar Days) | Primary Driver of Variation |
| Administrative Review | 15 Days | Missing mandatory eSTAR documents |
| Substantive Review | 60 Days | Complexity of software/cyber analysis |
| Decision Window | 90 Days (Total) | “Stop-Clock” due to AI Requests |
| Post-AI Response | N/A | Quality of technical response |
The FDA 510k timeline and process start long before the 510k submission is submitted to the FDA Customer Collaboration Portal (CCP). The eSTAR interactive PDF template, with its strict data-driven template, is the new backdrop for all submissions.
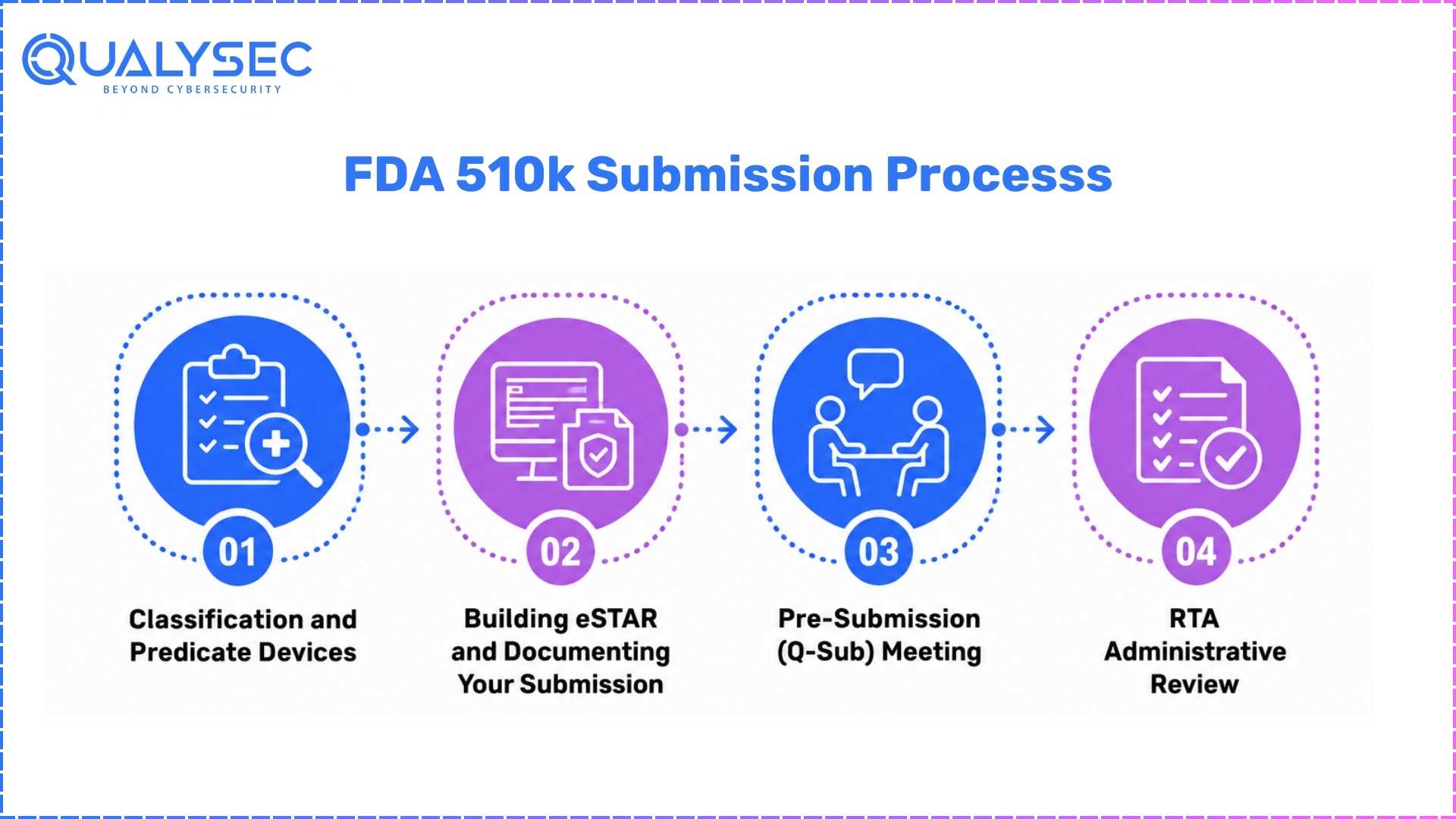
Step 1 – Classification and Predicate Devices
This is one of the most important steps because the wrong classification or weak predicate choice can delay the entire submission. You should correctly classify your device (as Class I, II, or III) according to the FDA’s classification database. For 510(k) submissions, you will most likely be working with a Class II device. Your selection of a predicate device – a device legal to market to which you compare your device for substantial equivalence – is a pivotal moment. If your predicate is old (e.g., cleared 15 years ago), there is a risk the FDA will request new performance data that goes beyond the predicate’s original data.
Step 2 – Building eSTAR and Helping to Document Your Submission
eSTAR is more than a basic electronic form. It is an interactive template that guides the submitter through required content based on device type, intended use, software, cybersecurity, and other factors. You must fill in the summaries in the Design History File (DHF), which includes:
- Design Inputs & Outputs – Demonstrating the device is designed for its intended purpose.
- Risk Analysis (ISO 14971) – Detailed listing of all potential hazards, the actions taken, and residual risk analysis.
- Performance Testing – Bench, animal, or clinical testing. Bench testing in 2026 is often required to meet clinical standards.
Step 3 – Pre-Submission (Q-Sub) Meeting
The Pre-Sub process is the new fast track, but it is optional. When you send your test protocols to the FDA for review and feedback, prior to the final tests, you receive a letter of agreement. Following FDA feedback from a Pre-Submission meeting can reduce the risk of unexpected questions later, although the FDA may still request additional information if new issues arise.
Step 4 – RTA Administrative Review
After you submit your package, it will be subject to an acceptance review. This is a go/no-go review of the Refuse to Accept (RTA) list. This is where the FDA looks for the missing signatures, incomplete cybersecurity information (such as the SBOM), and is the time when they look for incorrect labeling. Failure to meet FDA cybersecurity guidelines can also increase the risk of a Refuse-to-Accept (RTA) decision.
Contact Today to Avail Qualysec’s 3-Layered Approach to Ensure Your Software is 2026 FDA Ready!
Need a compliance-ready? Contact us Today
Timelines – MDUFA Targets vs 2026 Reality
The FDA 510k timeline is often misunderstood to be a 90-day clock. The reality is that the 90-day time frame is the FDA’s internal goal for its active review time. This figure doesn’t include time that is stopped while you are responding to Additional Information (AI) requests.
While FDA review goals are often discussed around 90 FDA days, real calendar timelines can stretch when the review clock stops for RTA responses or Additional Information requests.
| Delay Category | Root Cause | 2026 Mitigation Strategy |
| Cybersecurity | Missing SBOM/Threat Model | Include proactive VEX reports and automated scan summaries. |
| Traceability | Disconnect between Risk and Testing | Use a centralized digital DHF to link every requirement to a test. |
| Technical Parity | Inconsistent Predicate comparison | Use a side-by-side comparison table to explicitly address all “new” features. |
| Human Factors | Poor usability testing data | Conduct formative evaluations before the final summative submission. |
The Clock Stop Mechanics
The AI Request (Additional Information) is the biggest factor affecting your TTD. The lead reviewer issues an AI letter when they believe there is an issue with your technical documentation. At this point, the FDA’s 90-day clock stops. You will have 180 days to respond. If you rush the response and/or do not fully address the FDA’s concerns, you may receive a second AI request, and so on, leading to a delay of 6-8 months.
An FDA Deficiency Letter, an Additional Information (AI) request, identifies gaps in the submission that must be addressed before the review can move forward.
Manufacturers who consistently achieve the 120-day goal (the Gold Standard for 2026) are those who respond to the first AI request as though their application will be reviewed for any weaknesses. They answer the questions posed by the AI in their initial eSTAR submission by supplying them with an answer supported by a bulletproof technical justification.
Proactive Strategy for 2026
The 2026 regulatory requirements are a call to arms from the old submit and wait approach. Since the Quality Management System Regulation (QMSR) has been fully in effect with ISO 13485:2016 as of February 2, 2026, the FDA 510k timeline is no longer a discrete event but an ongoing process in your quality management system. Today, the best way to be proactive is to have a focus on being ready for the Total Product Life Cycle (TPLC) and documenting all design decisions to the level of detail required by the eSTAR format.
In this high-stakes environment, the most successful manufacturers are taking the opportunity to use Pre-Submission (Q-Sub) meetings six months before their intended filing date. This strategy enables you to establish your predicate and clinical testing plans with the FDA, thereby eliminating the possibility of a stop-clock Additional Information (AI) request during the actual submission review. For AI-enabled devices, manufacturers should evaluate whether a Predetermined Change Control Plan may help support planned future model updates.
Lastly, proactive compliance means that cybersecurity is prioritised as a fundamental safety attribute. By applying a Secure Product Development Framework (SPDF) early in the design process, your threat model, Software Bill of Materials (SBOMs), and pen test reports are completed well before the first line of code. And by aligning your technical prowess with these regulatory changes, you turn the FDA 510k timeline into a reliable, efficient vehicle for delivering your product to market.
Qualysec Technologies – A Line of Defense
The aim of the FDA 510(k) process that most companies struggle with is the technical documentation phase. This is where Qualysec Technologies’ expertise and capabilities come into play. Qualysec is a premier, next-generation cybersecurity company. They specialise in deep-testing or human-led, AI-powered services and solutions to address high-risk requirements of the medical and regulatory industries, rather than just consulting.
Qualysec has a three-layered defence system. In the 2026 world, where the FDA often rejects submissions due to inadequate cybersecurity information, Qualysec’s strategy is to leave nothing behind. They offer the human intuition that the FDA is seeking in a Threat Modeling and Pentest report.
Three-Layered Defence System – How it Works
- Layer 1 – Scanners: Employing state-of-the-art scanners to detect the low-hanging fruit and common vulnerabilities in the devices’ firmware and web interfaces.
- Layer 2 – AI Driven: Sophisticated machine learning algorithms are used to detect the traffic and code patterns to identify anomalies and complex attacks that scanners fail to detect.
- Layer 3 – Human Powered: Qualysec’s top researchers manually try to hack the device, mimicking techniques that hackers use to hijack medical devices. This gives the critical human intuition to the report, making a future-ready submission.
Through their dashboard, Qualysec gives manufacturers visibility into their project, seeing their security gains at each step of the way. They help solve the speed vs. accuracy challenge and keep you on schedule by offering optimum speed with critical human intuition.
Conclusion
To manage the FDA 510k timeline in 2026, it is suggested to move away from a check-the-box approach and adopt a technical excellence approach. The FDA’s requirement of mandatory eSTAR submissions and the increased scrutiny of cybersecurity issues mean that the technical aspects of your submission are the most important component in the time to market. Recognising the RTA period, the substantive interaction, and the risks of poor documentation when it comes to software, manufacturers can sail through the regulatory process.
Talk to Qualysec Technologies for the Security Testing!
Consult with our cybersecurity experts
Discuss your unique security requirements and discover how we can help your business.
FAQs
Q.Does a 510k expire?
The 510(k) clearance has no specific expiry date. Once the FDA has deemed your device to be substantially equivalent to a predicate, your 510(k) will never expire. But if you make substantial changes to the device’s intended use, design, or software that might impact its safety or effectiveness, you are legally obliged to file a new 510(k) to remain compliant and continue to market the device.
Q.What are the FDA 510k rules?
The main rules are found at 21 CFR Part 807, Subpart E. These regulations require companies to give the FDA at least 90 days’ notice before selling a device. The application should include technical comparisons to a “predicate” device, evidence of clinical and non-clinical safety, and robust cybersecurity evidence, such as SBOMs and threat models, to demonstrate that the device is as safe as those already on the market.
Q.What is the average review time for 510k?
Theoretically, the FDA aims to make a decision under MDUFA IV/V in 90 calendar days, but the average review time based on recent reports has been 140-170 days. This is typically because of “AI Requests” (Additional Information), which put the FDA on hold. If you lack complete documentation or detailed information on cybersecurity testing, the process could easily take six months or more as you work to resolve these technical issues.
Q.What are the 5 phases of FDA 510(k) clearance?
This includes:
- Classification, to determine if your device is Class I, II, or III
- Predicate Selection, to find a legally marketed device
- Data Gathering, to perform bench, software, and clinical testing
- Submission & Review, to navigate the 15-day RTA and 60-day substantive interaction periods
- Final Decision to receive the “Substantially Equivalent” letter so that you can commercially market the device in the United States.
Q.What is the timeline for FDA 510(k) clearance?
Typically, the FDA 510k timeline is 4 to 6 months for Class II devices. This includes the first 15 days of initial administrative review and the substantive review, which concludes around day 90. But the FDA 510(k) timeline for high-risk medical devices that require a Premarket Approval (PMA) can take longer – 12 to 24 months – as the device undergoes extensive human clinical trials and more rigorous scientific reviews by the CDRH.








































































































































































































































































































































































































































































































































































































































































































0 Comments