






Introduction
FDA readiness means a continuous state of preparation for the USA Food and Drug Administration (FDA) inspection. It is a mandatory requirement for all kinds of organisations that are involved in manufacturing, processing, storage, distribution, or testing of FDA-regulated products, including pharmaceuticals, biologics, medical devices, dietary supplements, food, and cosmetics. Non-compliance can lead to Form FDA 483 observations, warning letters, product holds, recalls, or legal enforcement actions by regulatory authorities.
In this guide, we will understand what an FDA inspection is, organisations subject to FDA inspection, its types, cybersecurity requirements under the FDA inspection, and a checklist for your reference.
What is an FDA inspection?
FDA inspection is an official, unannounced (often) inspection conducted by the U.S. Food and Drug Administration (FDA) to check whether a facility and its operations comply with the U.S. Food and Drug Administration (.gov) regulations. The main aim of inspection is to verify safety, quality, and manufacturing standards (cGMP).
FDA inspection readiness means your organization has appropriate systems, policies, documentation, people, processes, and supporting technologies ready for inspection at any time by FDA investigators.
Types of FDA inspection
- Pre-Approval Inspections (PAI) – Occur once the company submits an application to market a new product that falls within the eligible category. The FDA inspects to verify the application data and confirm whether the facility is capable of manufacturing the specified product.
- Routine surveillance inspection – Regularly scheduled inspections are conducted to evaluate whether the manufacturer is maintaining quality standards and following its own Standard Operating Procedures (SOPs).
- For cause inspection – Triggered by consumer complaints, whistleblowers, product recalls, and severe health cases, including death. This type of inspection is different from the others, as investigators will dig deeply into the system and processes and address immediate and long-term concerns.
- A compliance follow-up inspection was carried out by the FDA to verify whether the previously identified problems had been addressed. If corrections are not implemented, the FDA is authorised to take enforcement actions.
QMSR Implementation
Quality Management System Regulation (QMSR) is a system that defines how a company delivers products or services that meet quality standards.
Applicability: QMSR applies to the finished device manufacturers that wish to commercially distribute medical devices in the USA.
QMSR incorporates:
- ISO 13485:201
- Medical devices – Quality management systems
- Clause 3 of ISO 9000:2015
Core Requirements Under the FDA Quality Management System Regulation (QMSR)
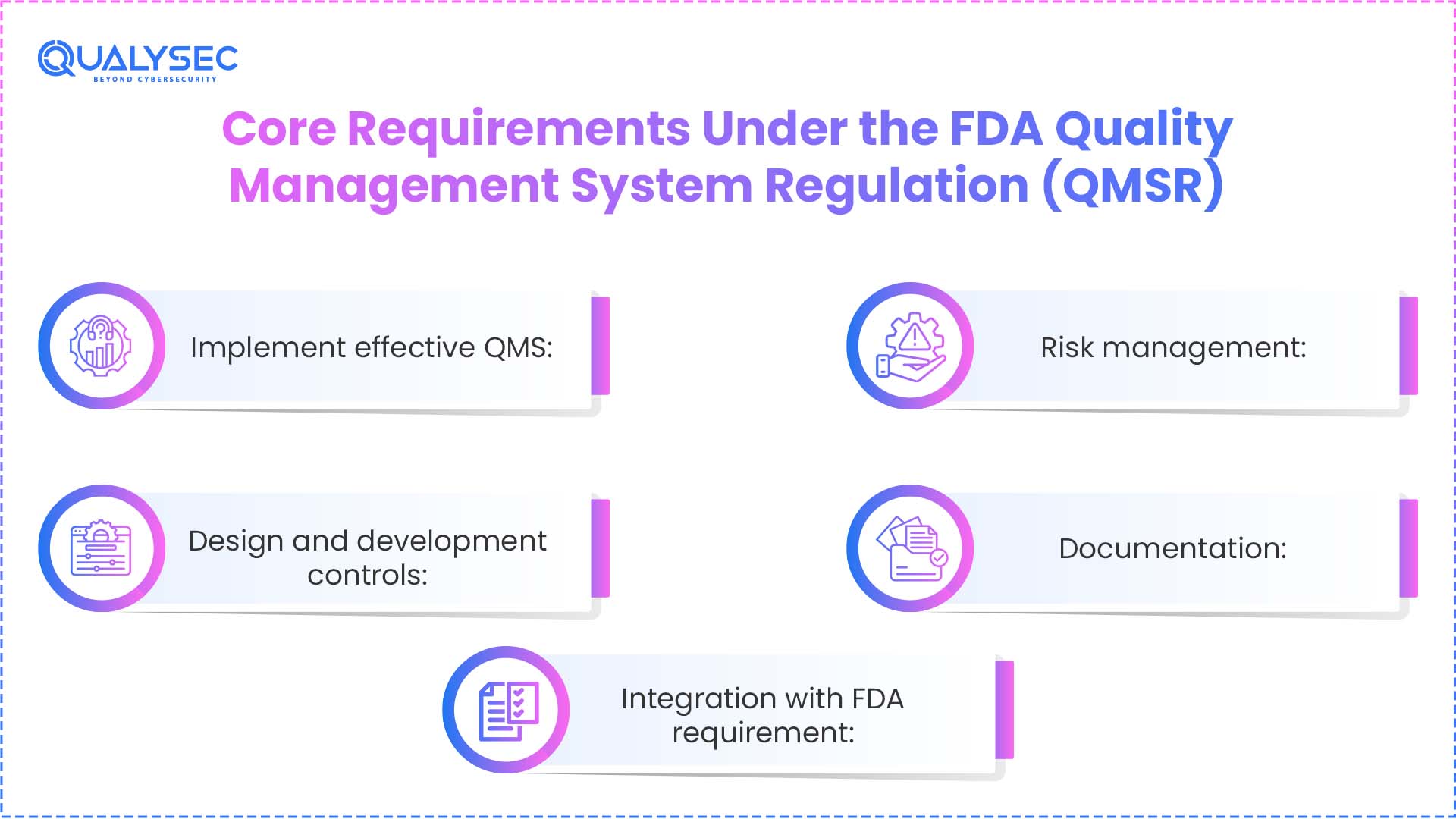
1. Implement effective QMS: Manufacturers of finished medical devices are required to set up, document, implement, and maintain a Quality Management System (QMS) across the entire product lifecycle.
2. Risk management: The eligible entities are mandated to:
- Identify and evaluate risks associated with devices and processes
- Implement appropriate risk control measures
- Monitor residual risks and take corrective actions when needed
3. Design and development controls: If the company/firm designs medical devices, QMSR requires the company to:
- Plan how the device will be designed
- Clearly define user needs and regulatory requirements
- Test and verify that the design works as needed
- Validate that the final device is suitable for real-world use
- Control and document any design changes
4. During the inspection, the FDA checks:
User needs → design requirements → testing → risk controls → final product
5. Documentation: QMSR requires companies to properly manage all quality-related documents and records. Organisations must ensure that they properly approve documents before use, remove old or obsolete documents, keep records complete and accurate, store them properly, and ensure that records are properly retained.
. FDA inspectors can review internal audit reports and management reports
6. Integration with FDA requirement: Even though the QMS system aligns with the ISO 13485, manufacturers have to comply with the existing FDA regulations, which are:
- Unique Device Identification (UDI) – 21 CFR Part 830
- Medical Device Reporting (MDR) – 21 CFR Part 803
- Corrections and Removals – 21 CFR Part 806
- Device Tracking – 21 CFR Part 821, where applicable
Cybersecurity requirements within a Quality Management System (QMS)
For medical device manufacturers, cybersecurity within a Quality Management System (QMS) extends beyond data integrity controls and includes product security, risk management, and regulatory compliance throughout the device lifecycle. It requires:
- Cybersecurity risk-based management is integrated into design controls and risk management processes.
- Secure product development practices, including threat modeling, secure coding, vulnerability assessment, and penetration testing where appropriate.
- Access controls ensure that systems and device software are limited to authorized individuals.
- Audit trails that capture the creation, modification, and deletion of electronic records.
- Protection of electronic records against accidental or malicious alteration, deletion, or loss.
- Secure configuration management and formal change control for patches, updates, and cybersecurity modifications.
- Monitoring, identification, reporting, investigation, and timely remediation of cybersecurity vulnerabilities and incidents.
- Postmarket surveillance and coordinated vulnerability disclosure processes.
- Corrective and preventive action (CAPA) processes linked to cybersecurity incidents and risk reassessment.
- Data confidentiality, integrity, and availability safeguards, including validated backup and disaster recovery mechanisms to ensure business continuity.
FDA cybersecurity requirements? Schedule a security assessment with our Qualysec experts today!




































































































































































































































































































































































































































































































































































































































































































0 Comments