







Regulatory framework for BIMO Inspections
| Regulation / Guidance | It applies |
| 21 CFR Part 50 | Investigators, Sponsors |
| 21 CFR Part 54 | Investigators, Sponsors |
| 21 CFR Part 56 | IRBs / Ethics Committees |
| 21 CFR Part 312 | Sponsors, Investigators, CROs |
| 21 CFR Part 812 | Medical Device Sponsors, Investigators |
| 21 CFR Part 11 | Sponsors, CROs, Sites, Vendors |
| ICH E6 (GCP) | All trial stakeholders |
| FDA Compliance Program Guidance Manuals (CPGMs) | FDA Inspectors (publicly available) |
| FDA Guidance on Risk-Based Monitoring | Sponsors, CROs |
Requirements Under BIMO Inspection
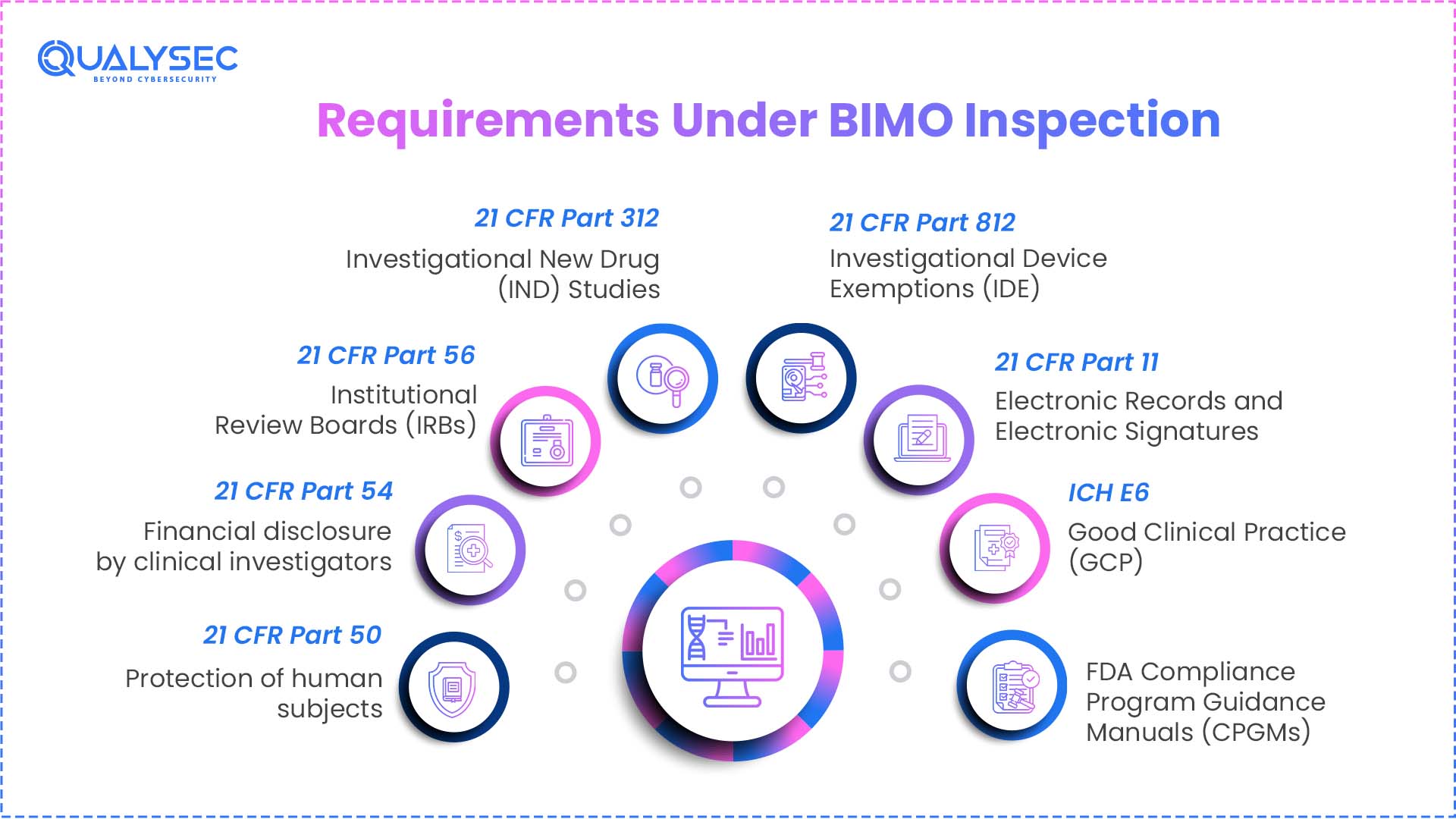
1.21 CFR Part 50 – Protection of human subjects:
- Subjects must be informed of the study purpose, procedures, risks, benefits, and alternative treatments.
- The FDA requires that informed consent be obtained before a subject participates in a clinical investigation, unless an exception applies.
- Consent must be voluntary, legally effective, and written in language understandable to the subject.
- The document consent must be reviewed and approved by an IRB prior to use.
- During the BIMO inspections, FDA shall verify the timeliness, completeness, and consistency of the consent process.
2.21 CFR Part 54 – Financial disclosure by clinical investigators:
During BIMO inspections, the FDA assesses whether researchers identified, properly documented, and managed financial conflicts to protect data integrity.
FDA requires clinical investigators to disclose financial interests, such as compensation arrangements, proprietary interests, and significant equity holdings, that could affect the objectives of the study.
- Sponsors must collect, review, and maintain accurate financial disclosure information.
- Sponsors must submit certifications or disclosures to the FDA.
- Financial information must be updated during the study and for one year following study completion.
3.21 CFR Part 56 – Institutional Review Boards (IRBs):
- During BIMO inspections, the FDA evaluates IRB operations, documentation, and the effectiveness of oversight.
The FDA requires IRBs to be properly constituted to review and approve clinical protocols and informed consent documents before the initiation of the study.
- IRB shall review records, meeting minutes, and approval documentation at appropriate intervals.
4.21 CFR Part 312 – Investigational New Drug (IND) Studies:
- During BIMO inspections, the DA focuses on protocol, FDA compliance, safety oversight, and data reliability.
- The FDA requires sponsors to conduct drug trials in accordance with the approved protocol and IND regulations.
- Investigators must properly store, control, and account for investigational drugs, and they must maintain accurate case histories and source records.
- In the event of adverse events, safety information must be reported in accordance with regulatory timelines.
5.21 CFR Part 812 – Investigational Device Exemptions (IDE):
- During BIMO inspections, the FDA evaluates device accountability, safety reporting, and study conduct.
FDA requires device studies to follow the approved investigational plan and IDE conditions, and the devices to be properly controlled, stored, and accounted for. - In the case of any unanticipated device, adverse device effects must be properly reported.
6.21 CFR Part 11 – Electronic Records and Electronic Signatures:
During BIMO inspections, the FDA assesses data integrity and system controls, and requires electronic systems used in clinical trials to be validated for their intended use.
- Therefore, access to electronic records must be limited to authorized individuals only.
- Secure, computer-generated audit trails must track record, modification, and deletion.
- Electronic signatures must be unique to an individual and legally binding.
- And the records must be protected from unauthorised alteration.
7. ICH E6 – Good Clinical Practice (GCP):
- During BIMO inspections, the FDA uses ICH E6 to assess ethical conduct and data credibility, and the FDA expects clinical trials to follow Good Clinical Practice principles.
Documented and protected, critical data and processes that affect the safety of the subject.
Trials must follow the approved protocols and regulatory requirements, with appropriate monitoring and quality management systems in place.
8. FDA Compliance Program Guidance Manuals (CPGMs):
- FDA uses CPGMs to define how BIMO inspections are planned and conducted. The manuals outline the inspection scope, objectives, and methods for collecting evidence.
- The manual guides inspectors on evaluating investigators, sponsors, CROs, and IRBs.
- CPGMs support consistent regulatory decision-making across inspections.
Risk-Based Monitoring on FDA Guidance
- FDA expects sponsors to implement monitoring strategies based on study-specific risks.
- Critical data and processes that affect the safety of the subject must be documented and protected.
- Centralised and remote monitoring may be used alongside targeted on-site visits.
- Risk assessments must be ongoing and appropriately documented.
FDA may conduct BIMO inspections:
- For cause, based on complaints, adverse events, or data anomalies
- Routine surveillance, during an ongoing or completed study
- Application-driven, during the review of INDs, NDAs, BLAs, or PMAs
Sponsor Accountability:
The FDA emphasises that ultimate responsibility for trial compliance rests with the sponsor. While sponsors may delegate trial activities to CROs, vendors, or sites, they cannot delegate accountability. The sponsors are expected to:
- Maintain effective watch over CROs and vendors
- Implement appropriate monitoring and quality management systems
- Ensure timely detection and correction of noncompliance
- Keep clear and concise documentation of decisions, risk management, and actions taken
Any failure to properly oversee vendors is one of the most common reasons for receiving Form 483 observations.
Understanding FDA Form 483
Form 483 is a tool for communicating potential regulatory violations observed during a BIMO inspection. It is an early warning that highlights areas where a company’s practices may not fully comply with FDA regulations. The FDA issued the form at the conclusion of an inspection and lists specific observations, such as inadequate management, poor documentation, or gaps in quality management systems. Companies that receive a Form FDA 483 must investigate the root cause of each observation and submit a written response that specifically outlines corrective and preventive actions (CAPA) and timelines for resolution.
Common BIMO Inspection Deficiencies
A.Investigator and Site-Level Deficiencies
- Protocol deviations not adequately documented or justified
- Failure to follow the investigational plan or signed agreements
- Inadequate or missing informed consent documentation
- Delayed or incomplete adverse event reporting
- Poor investigational product accountability
- Inadequate communication with the IRB
- Misrepresentation of investigational products as safe or effective
B. Sponsor, CRO, and Monitor Deficiencies
- Inadequate monitoring plans or execution
- Failure to identify or correct investigator noncompliance
- Weak investigational product supply chain controls
- Study initiation without required FDA or IRB approvals
- Insufficient oversight of electronic systems and vendors
Qualysec: Your Trusted Partner
FDA BIMO inspections are a mandatory requirement, it examine electronic systems, data integrity, and vendor oversight. Organisations must show that their clinical trial data is secure, reliable, and trustworthy. Qualysec supports sponsors, CROs, and clinical research organisations.
- Qualysec helps organisations identify vulnerabilities in clinical trial applications, electronic data capture (EDC) systems, eTMF platforms, and laboratory information systems during a BIMO inspection.
- During BIMO inspections, FDA investigators frequently assess compliance with 21 CFR Part 11, including system validation, access controls, audit trails, and protection against unauthorised data modification. Qualysec’s assessments help verify that these controls are effectively implemented.
- Sponsor accountability remains the focus of the BIMO program. Even when trial activities are outsourced, sponsorsremain responsibility for oversight of CROs, vendors, and technology providers. Qualysec supports this surveillance through independent security evaluations of third-party systems.
- Qualysec helps organisations reduce inspection risk, prevent data integrity-related observations, and maintain confidence in clinical data submitted to the FDA.



































































































































































































































































































































































































































































































































































































































































































0 Comments