






The FDA 510k submission guidance in 2026 is more relevant than ever to the medical device manufacturing industry worldwide. Approximately 3,200 510(k) submissions are reviewed annually by the FDA. In 2025, more than 3,238 clearances were granted, with a significant portion of these applications originating from over 36 countries. It is a very dynamic market because it is highly regulated and it is based on technological innovation, especially in the digital health and connected devices field, which has to comply with new cybersecurity standards. Newer revisions to the FDA 510k guidance and regulations are in, most notably the official transition to the Quality Management System Regulation (QMSR), which aligns FDA requirements with ISO 13485:2016 as of February 2, 2026.
These revisions highlight the use of the eSTAR submission format as mandatory and increase the level of cybersecurity requirements across the entire lifecycle of devices, which leads to an increased relevance of effective risk management methods. The successful FDA 510k submission guidance requires manufacturers to incorporate these emerging technical and regulatory factors —including the new QMSR framework— early in the design stage of their products to maximize clearance and successful market entry.
Need help with FDA 510(k) cybersecurity compliance? Connect with Qualysec experts today and get end-to-end guidance for your medical device approval.
Cybersecurity Compliance with the FDA
The U.S. Food and Drug Administration (FDA) has been focusing more on the cybersecurity of medical devices, especially the 510(k) premarket notification process. The most recent FDA 510k guidance, issued in February 2026, has set very stringent conditions. These conditions must be incorporated at the initial stages of device development and submission to the FDA to guarantee patient safety, data integrity, and regulatory approval. Cybersecurity compliance is no longer a luxury. It is now an essential component of the FDA 510 k guidance framework that requires a lifecycle-based security approach.
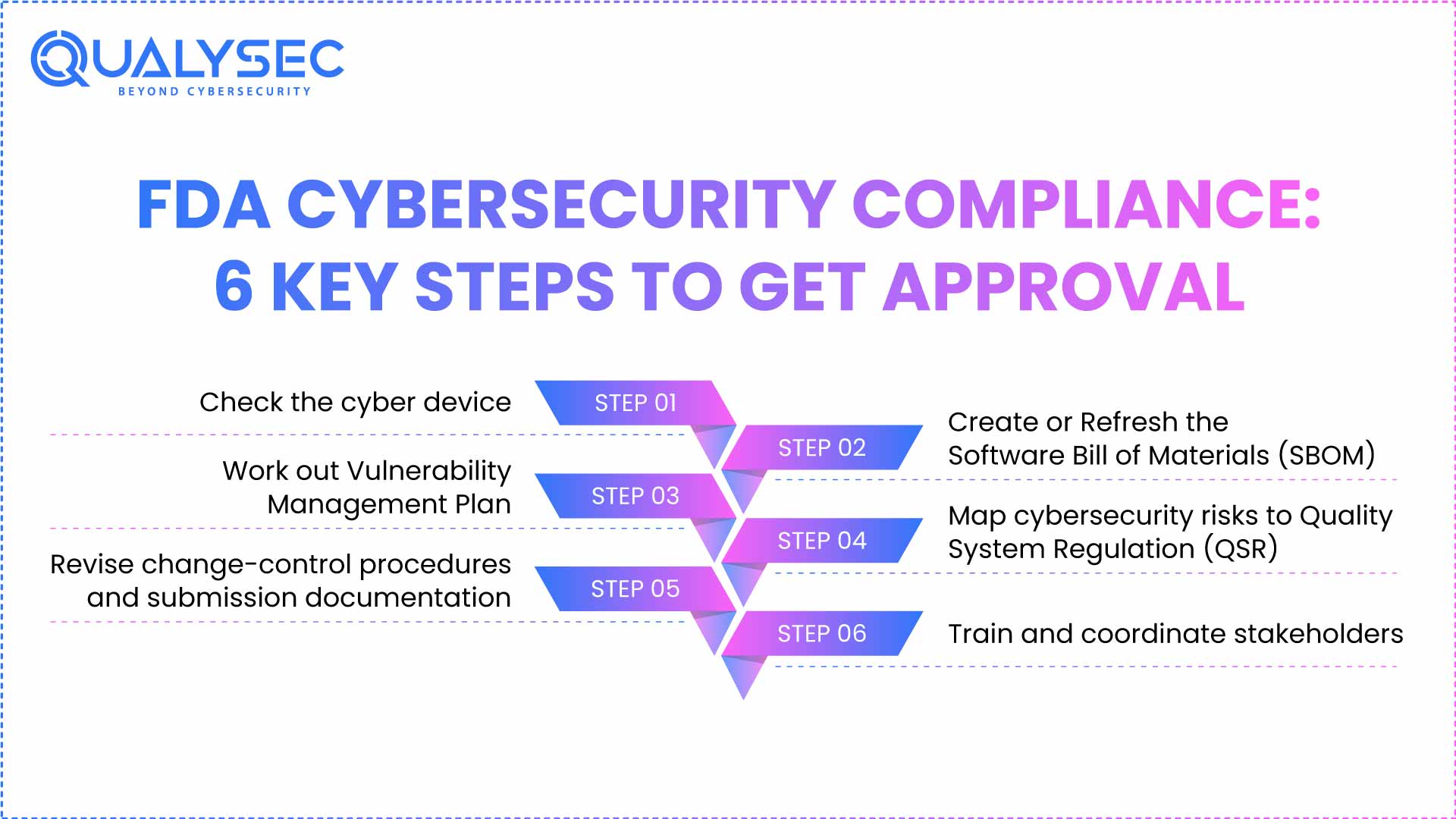
1st Step – Check Cyber Device
- Cybersecurity does not apply to every device. First, decide whether the FDA should consider the device a cyber device.
- The most recent FDA cybersecurity guidance, issued in February 2026, sets the current expectation for all premarket submissions. Section 524B(c) of the FD&C Act defines any device that has software (embedded or standalone) and network connectivity (including latent modules such as debug ports or wireless interfaces) as a cyber device.
- This designation initiates the entire cybersecurity documentation and submission requirements in accordance with FDA 510(k) directives.
2nd Step – Create or Refresh the Software Bill of Materials (SBOM)
- A component of FDA cybersecurity compliance that is very important is the SBOM.
- Manufacturers are required to create a complete, machine-readable SBOM that describes all software components, both open-source libraries and commercial third-party code, as well as proprietary modules.
- This report should be kept in chronological order throughout the life cycle of the device and should adhere to the standards of the National Telecommunications and Information Administration (NTIA).
- SBOM enables tracking of vulnerabilities and risk reduction activities, which are key to the new submission requirements at FDA.
3rd Step – Work out Vulnerability Management Plan
- A formalized vulnerability management strategy has become a new mandatory requirement.
- This plan should include receiving, triaging, remediating, and communicating vulnerabilities. Crucially, manufacturers must now implement a Secure Product Development Framework (SPDF). This includes: 1) Threat Modeling to identify risks before market entry, and 2) Detailed Security Architecture Views showing ‘trust boundaries’ and defenses against multi-patient harm.
- It also involves setting timelines when risks are to be addressed, when patch releases will be made, and through which channels will be made to provide transparency to the customer in the event of any threats being discovered.
- Teams should incorporate this process at an early stage to maintain compliance throughout the device’s pre-market submission and post-market monitoring.
4th Step – Map Cybersecurity Risk to Quality Management System Regulation (QMSR)
- In its 2026 guidance, the FDA provides express links between cybersecurity risk management and the Quality Management System Regulation (QMSR), which officially replaced the old QSR in February 2026. Manufacturers are required to indicate how their cybersecurity practices align with ISO 13485:2016.
- This is because manufacturers are required to indicate how their cybersecurity practices are aligned with the Secure Product Development Framework (SPDF) and risk management procedures under the new QMSR.
- This correspondence would make cybersecurity a quality and never-ending aspect of device development, production, and upkeep.
5th Step – Revision to Change-Control Procedures and Submission Documentation
Manufacturers must now report changes to devices that affect cybersecurity posture in a revised submission or amendment under the FDA’s change-impact taxonomy –
- Amendments that are likely to modify cybersecurity need an additional 510(k) or PMA.
- Minor or improbable impact changes will only necessitate an internal documentation change.
This taxonomy requires manufacturers to update change-control Standard Operating Procedures (SOPs) to support reporting promptly.
Also, you must include comprehensive cybersecurity documentation in the 510(k) submission packet through the latest Version 6.1+ of the FDA’s Electronic Submission Template and Resource (eSTAR). This updated template now includes integrated fields for QMSR compliance and specific cybersecurity architecture views.
6th Step – Train and Coordinate Stakeholders
- Compliance requires the coordination of R&D, Regulatory Affairs, Quality Assurance, DevOps, and suppliers.
- Continued training and awareness of the new cybersecurity requirements will make sure that all members of the team know their roles, duties, and schedules.
- The formation of cross-functional alignment will help to respond more easily and successfully react to cybersecurity incidents after market deployment.
Qualysec Technologies for FDA 510(k) Cybersecurity Compliance
Qualysec Technologies is one of the most successful cybersecurity companies offering a wide range of penetration testing, vulnerability testing, and compliance services that are referred to as FDA 510k guidance and regulations. We have found a niche in offering verified, process-based testing solutions to medical device manufacturers operating in the FDA 510(k) regulatory environment.
Proven Process-Based Testing
In contrast to most cybersecurity companies that use only manual or automated testing, Qualysec uses a combination of the two in a careful and data-driven process. The combination methodology will provide more detailed and credible vulnerability testing of security vulnerabilities that are material based on FDA 510k guidance and compliance regulations.
Domain Knowledge in Medical Devices
Qualysec has specialized in medical device cybersecurity and is deeply familiar with current FDA 510k submission guidance and lifecycle security requirements. This expertise makes the company an invaluable partner to a manufacturer that aims to capture the FDA market clearance.
Full-scale Compliance Support
In addition to penetration testing, Qualysec supports its clients with in-depth risk evaluation, software bill of materials (SBOM) verification, post-market monitoring plans, and documentation procedures that are part and parcel of FDA cybersecurity submissions. This whole support facilitates the torturous regulations.
Strong Track Record
Qualysec has already been able to fulfil more than 450 penetration testing and compliance engagements across the world, with a clean track record of zero data leakage reported among the clients. Regarding the reliability and confidence of regulatory preparedness and patient safety, the performance is high.
Customized Solutions
Each project will be driven by client insight about the type of technology in the device, their risk profile, and ambition in the market. Qualysec modifies its testing and guidance strategy to meet the requirements and maximize the effect of compliance resources.
Quality Client Support and Interaction
Qualysec likes doing business as a client support and interaction. Regular consulting and retesting provided by the company improve remediation and security posture over time.
Services Overview
FDA 510(k) Cybersecurity Compliance Consulting
QMSR & ISO 13485:2016 Gap Analysis
eSTAR Technical File Preparation (v6.1 Update)
Secure Product Development Framework (SPDF) Implementation
Confirm Authenticated Hybrid Penetration Testing (Manual + Automated)
In short, Qualysec uses a proven process-based testing strategy aligned with FDA 510k guidance and standards. This active approach combines the experience of human operators with the latest automated solutions and strict verification of data to offer a level of security assurance never seen before.
Ensure your medical device meets FDA 510(k) guidance without delays. Schedule a cybersecurity compliance assessment with Qualysec now!




































































































































































































































































































































































































































































































































































































































































































0 Comments