






FDA Cybersecurity Guidance You Must Align With
The FDA released cybersecurity guidance for medical devices in 2023. It explains what should be included in FDA 510(k) submissions from a cybersecurity perspective.
The focus is on building security into the product early. This includes:
- SPDF
- Risk-based controls
- SBOM
- Postmarket cybersecurity management
It also aligns with ISO 14971 for safety risk and AAMI TIR57 for cybersecurity.
Definition of a “Cyber Device” (Scope Clarification)
The FDA classifies a device as a cyber device based on a few clear conditions:
- It includes software
- It connects to a network or the internet
- It has potential vulnerabilities that could be exploited
If your device meets these criteria, it falls under cybersecurity requirements in Premarket Notification (510(k)) submissions.
Common examples include:
- Connected infusion pumps
- Remote patient monitoring systems
- AI-driven diagnostic tools
One important detail many teams overlook is scope. Even devices with limited connectivity, such as those updated through USB or other external methods, still fall under the category of cyber devices.
Secure Product Development Framework SPDF: The Core of FDA Evaluation
An SPDF is not a single document. It is how your team builds, tests, releases, and maintains a secure product from start to finish. The FDA looks at this as a continuous process that runs across the entire device lifecycle.
What FDA Expects Inside SPDF
Your SPDF should be part of your existing quality system. It should align with QMS requirements under 21 CFR Part 820 and reflect standards like ISO 13485. This applies across both premarket and postmarket stages, depending on your device.
To meet expectations, your process should clearly include:
- Security requirements defined early in the design phase
- Secure coding practices were followed during development
- Verification and validation activities linked directly to security controls
- A plan to continuously monitor and address vulnerabilities after release
What matters most is not just having these elements written down. The FDA looks at how well your process actually works in practice. They expect to see consistency, traceability, and clear evidence that security is built into every stage, not added later.
Need help building an FDA-ready SPDF? Contact our experts today!
Lifecycle View: Cybersecurity Across the Product Journey
The FDA wants to see how security is handled from the very beginning, not patched in later.
You need to cover the full journey of your device:
Design → Development → Validation → Deployment → Postmarket
And within that journey, your work should connect like this:
Threat identification → Risk assessment → Controls → Testing → Monitoring
Think of it as a flow your team actually follows:
Design
↓
Identify threats
↓
Assess risks
↓
Build with controls in place
↓
Test those controls
↓
Release the device
↓
Monitor and respond to issues
What usually causes problems is timing. If security shows up only during testing or just before submission, it becomes obvious. That is where many submissions get flagged or rejected.
Threat Modeling Requirements
1. System-Level Threat Modeling beyond just STRIDE
When the FDA asks for threat modeling, it is not looking for a quick checklist or a single method like STRIDE. It wants to see how you have analyzed the entire system around your device.
This means you need to cover everything that could introduce risk:
- Device hardware
- Software layers
- Network connections
- Cloud components
- Update mechanisms
Your submission should make it easy to understand how data moves and where things can go wrong. In most cases, this includes:
- Data flow diagrams that show how information travels through the system
- Clear trust boundaries between internal and external components
- Entry and exit points where the device interacts with users or other systems
What matters here is how clearly you have thought things through. The FDA should be able to see how your team identified risks across the whole system and addressed them before they turn into real problems.
2. Required Threat Model Components
Your threat model should break things down in a way that is easy to follow. If anyone is reviewing it should quickly understand how your system works and where it can be exposed.
This is how you can structure it:
- Architecture analysis
Show how different parts of your system connect and interact. This is usually supported by data flow diagrams that map communication clearly. - Attack vectors
List the ways your device could be targeted. Common examples include man-in-the-middle attacks, privilege escalation, and firmware tampering. - Entry points
Identify where someone could try to access the system. This could be APIs, open ports, or wireless interfaces. - Assets
Highlight what needs protection. This often includes patient data, login credentials, and device firmware. - Trust boundaries
Explain where trust levels change. For example, how your device interacts with cloud services or a hospital network.
If a reviewer has to guess where risks exist, your threat model is not doing its job.
Example: A simple example can help make this clear. If your device sends patient data to the cloud, one obvious risk is data being intercepted during transmission. To handle this, you would apply controls like TLS encryption along with certificate pinning to secure the connection. The FDA looks for this kind of practical thinking, where risks are tied directly to how the device works, not just listed as general possibilities.
Cybersecurity Risk Management
1. Dual Risk Framework Explained
Safety risk and cybersecurity risk management are handled in different ways, but in real scenarios, they overlap. Safety is managed using ISO 14971, while cybersecurity follows AAMI TIR57. The FDA wants to see how a cyber issue can turn into a safety problem. For example, if someone gains access to a device and changes how it works, that is no longer just a security issue. It directly affects patient safety. That link needs to be clearly written in your risk analysis instead of being treated as two separate things.
2. Critical Mapping Requirement Hidden FDA Trigger
Many submissions list risks, but stop there. That is where the problem starts. You need to connect the dots. Take a simple case. Someone gets unauthorized access to a device. That access allows them to change dosage settings. Now the device delivers the wrong amount, and the patient is at risk.
This kind of flow should be written clearly in your analysis. If the path from threat to harm is missing or vague, it becomes hard to understand what the real impact is.
Software Bill of Materials SBOM: More Than a Component List
A Software Bill of Materials, or SBOM, is a detailed inventory of every software component inside your device. This includes proprietary code, third-party libraries, and open source packages. For devices that fall under the cyber device category, the FDA expects an SBOM as part of cybersecurity documentation in Premarket Notification (510(k)) submissions and other submission types.
1. FDA SBOM Requirements
Your FDA SBOM Requirements need to be created in a format that tools can read, such as CycloneDX or SPDX. It should give a clear view of what your device is built on, including open source components, third-party libraries, and any firmware dependencies. The FDA uses this to understand your software stack without having to piece information together from different places.
2. Advanced SBOM Expectations Often Missed
Just listing the components is not enough. You must include the version, any known issues linked to it, and the supported status, whether it is still supported or already outdated. Without these details, the list does not tell much.
This is where the SBOM becomes useful. It lets you see if something inside your software could turn into a problem later.
3. Sample SBOM Structure Recommended Section
A simple SBOM entry should make it easy to see what is used and if anything needs attention. For example:
“component_name”: “OpenSSL”,
“version”: “1.1.1”,
“cve”: “CVE-2023-12345”
This kind of structure helps you keep track of known issues and link them directly to the components in your device. It also makes updates and reviews much easier later on.
Security Controls: Risk-Based Implementation
1. Core Control Categories Expected by FDA
Security controls should match the actual risks in your device, not just follow a checklist. The FDA looks at whether the controls make sense for how your system works.
Some areas are almost always reviewed:
- Authentication and authorization to control who can access the device
- Encryption to protect data, both stored and during transmission
- Secure update and patching process so issues can be fixed safely
- Logging and audit trails to track what is happening on the system
- Device hardening to reduce unnecessary exposure
- Secure boot and firmware integrity validation
- Device identity and authentication mechanisms
- Least privilege and role-based access enforcement
What matters is how these controls are applied in your device, not just whether they are mentioned.
2. Mapping Controls to Threats Critical Requirement
Each control should tie back to a real problem. If you add a control, there should be a clear reason behind it based on the risk.
For example, if there is a risk of unauthorized access, you might use multi-factor authentication along with role-based access control to limit who can do what. Missing connections make it hard to understand why the team added certain controls in the first place.
Security Testing Requirements (Why VAPT Alone is Insufficient)
1. FDA Cybersecurity Requirements for 510(k) Submissions
One type of testing does not cover everything. Vulnerability scanning identifies known issues, while penetration testing shows how those issues can be exploited in real situations. Alongside that, you must test security controls to confirm they work as intended. Abuse case testing adds another angle by showing how users might use the device in unintended ways.
2. Hidden Expectation: Evidence-Based Testing
Testing needs to match how you described risks earlier. If something is in your threat model, it should be tested in a way that reflects real use, not just theory.
What you include as proof matters just as much. Screenshots, logs, and clear steps showing how an issue was found or used all help make your testing easier to follow. A short summary on its own does not give enough clarity.
Postmarket Cybersecurity Defined During Premarket Submission
Required Postmarket Plan: Your submission must explain how you will monitor the device and manage data release, as the product and consumers begin using it.
Vulnerability monitoring:
Explain how you stay aware of new issues. This could be through your own checks, feedback from users, or information coming from outside sources.
Patch management:
Describe how fixes are handled. Include how updates are prepared and how quickly they are pushed when a problem is found.
Coordinated vulnerability disclosure:
Make it clear how someone can report an issue to you. Also, mention what you do once you receive that report and how you handle the communication.
Incident response:
Explain what your team does when a security issue is confirmed. Walk through how you deal with it and fix it. Also, how do you reduce the chances of it happening again?
Full Traceability Model
End-to-End Traceability Example
Your documentation should connect everything in one flow. The FDA looks for clear traceability between the threat model, risk assessment, controls, and testing, all the way to what risk remains at the end. A simple way to write it is like this:
Threat: MITM attack
Risk: data gets changed during transmission
Control: TLS encryption
Test: simulate network interception
Residual risk: low
This kind of flow makes it easy to follow what you found, what you did about it, and what is still left after all controls are in place.
Why Traceability Drives FDA Approval
FDA Approval reviewers check if everything lines up. They look for three things. Is anything missing? Does it make sense from start to end, and is there proof for what you are claiming? If a risk is written, they check the control. A control is written, and they check the test. If the test is there, they look for the result.
FDA guidance clearly mentions that system elements, requirements, and testing should be traceable to each other.
If those links are clear, the review moves forward. If not, it creates confusion and slows things down.
What FDA Reviewers Look For” regarding “eSTAR Version 6.1+ Compliance.”
eSTAR Version 6.1 pushes everything into a fixed structure, so reviewers can quickly see if something is missing or does not line up. The template itself is designed to match how they review submissions, so if a section is incomplete, it becomes obvious right away.
If you include data from sources like health records or registries, you must state the source and the collection method. The FDA checks whether you can actually use the data to judge safety and effectiveness.
Since everything follows a standard format, reviewers can more easily compare one section with another. If something does not match or feels inconsistent, it stands out quickly during review. The structure also makes it easier to scan patterns across submissions, which is one of the reasons the FDA uses eSTAR in the first place.
Common Reasons for 510(k) Cybersecurity Rejection (RTA Triggers)
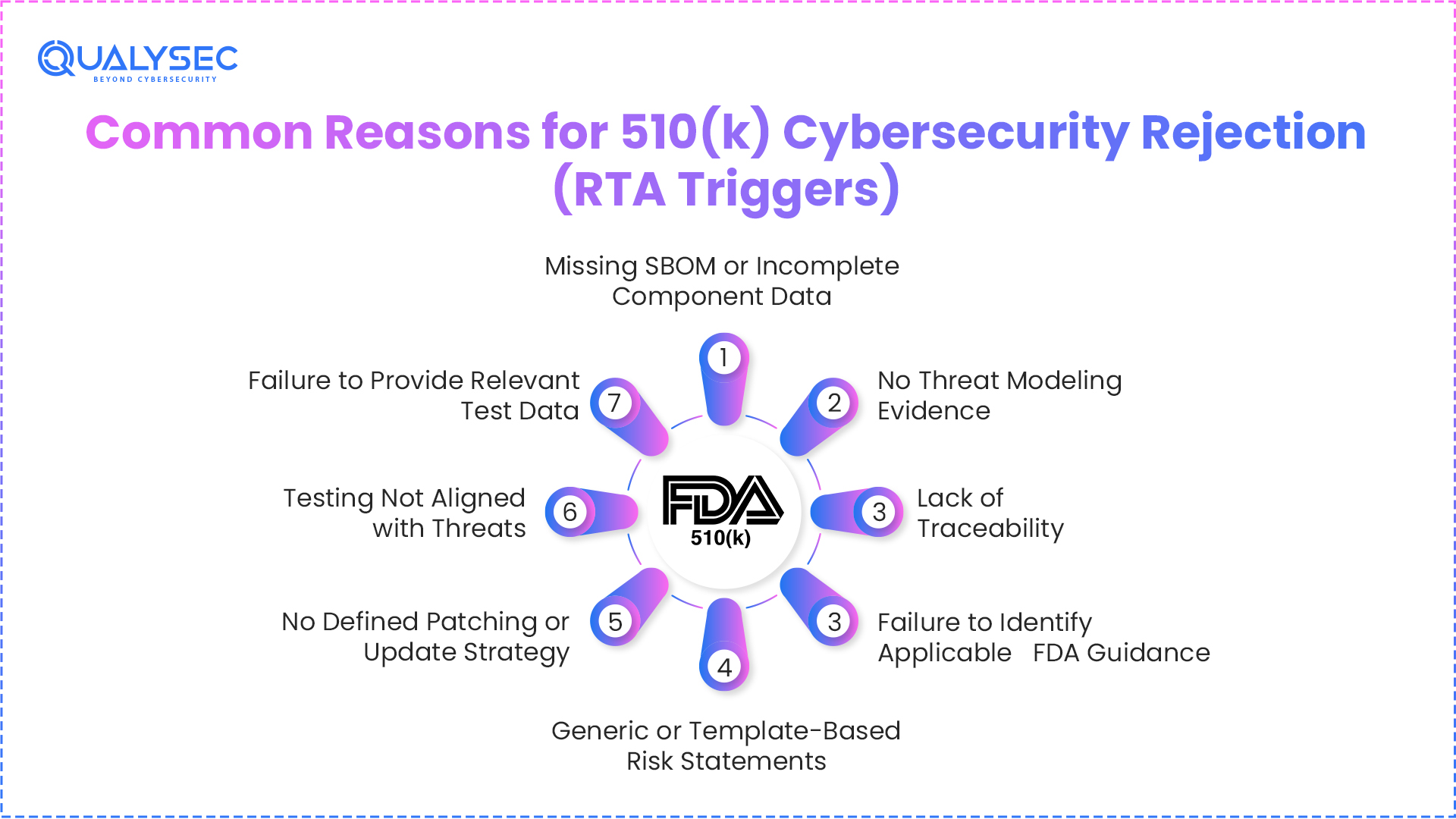
Missing SBOM or Incomplete Component Data
If your SBOM is missing or does not list all components properly, it creates a gap. FDA expects an SBOM as a core part of cybersecurity documentation.
No Threat Modeling Evidence
Some submissions mention risks but do not explain how they were identified. Without threat modeling, there is no clear view of how the system was analyzed.
Lack of Traceability
When risks, controls, and testing are not connected, the flow breaks. Reviewers cannot follow how one step leads to another, which raises concerns.
Failure to Identify Applicable FDA Guidance
Each device falls under specific guidance based on its product code. If you do not mention which guidance applies, the submission is treated as incomplete.
Generic or Template-Based Risk Statements
Risk sections that look copied or too broad do not help. They should reflect how your device actually works and where it can fail.
No Defined Patching or Update Strategy
Cybersecurity does not stop at submission. If there is no clear plan for handling updates and vulnerabilities after release, it becomes a gap in your documentation.
Testing Not Aligned with Threats
Testing should relate to the risks you identified earlier. If there is no connection between the two, reviewers cannot see why you performed certain tests.
Failure to Provide Relevant Test Data
Most 510(k) submissions need supporting test data, whether it is for software, safety, or performance. This includes areas like electrical safety, EMC, biocompatibility, or cybersecurity-related validation. Missing this kind of evidence can stop the submission from moving forward.
How Qualysec Helps You Meet FDA 510(k) Cybersecurity Requirements
Most teams already have security work in place. The problem shows up when you have to turn that work into something that fits a 510(k) submission.
That gap is where Qualysec focuses.
- Testing aligned with FDA review expectations
Testing is planned around how submissions are reviewed. This means mapping findings back to risks, controls, and evidence, not just listing vulnerabilities. - Covers the full device environment
Instead of testing one layer, Qualysec looks at APIs, cloud, and connected components together. This helps surface issues that only appear when systems interact. - Real attack paths, not just tool output
Manual testing combines with automated checks to reflect how an actual attack would play out, not just what scanners report. - Documentation that fits directly into submissions
Reports are written to support regulatory use. Each finding includes reproduction steps, maps to risk, and provides clear fixes so it can be used in 510(k) documentation without rework. - Compliance and mapping support
Findings aligned with standards like OWASP, NIST, HIPAA, and ISO 27001, making it easier to connect technical work with compliance requirements. - Support beyond the report
Retesting and follow-ups help make sure fixes are complete and properly documented before submission
Conclusion
The FDA does not focus on what you say you have implemented. It looks at what you can prove.
All cybersecurity controls in Premarket Notification (510(k)) Submission ultimately support the FDA’s requirement to demonstrate reasonable assurance of safety and effectiveness. A security issue is only relevant if it can impact how safely or effectively the device performs.
If you mentioned a control, you should include a test behind it. If you list a risk, you should provide a clear path showing how you handled it. This is what recent guidance keeps pointing to, linking risks, controls, and testing across the device lifecycle.
When these pieces connect, the submission is easier to go through. When they do not, reviewers have to stop and figure out what is missing. At this stage, cybersecurity shapes how the device is built. It is not something you add later while preparing documents.
Schedule a meeting with our experts to secure your Premarket Notification (510(k)) Submission FDA compliance.




































































































































































































































































































































































































































































































































































































































































































0 Comments