






Key Takeaways
- The FDA’s QMSR (ISO 13485:2016) transition is now fully effective as of February 2, 2026. Qualysec provides the mandatory gap analysis and auditing required to ensure your legacy QMS meets these new enforcement standards.
- Any model change falling outside the scope of an authorized PCCP will trigger a new 510(k) submission; for adaptive AI products, a robust PCCP is now a critical commercial requirement to maintain market relevance.
- The 2025 FDA guidance on cybersecurity, updated June 2025, required mandatory use of SBOM and pen-tests on devices that had a latent connection, such as USB ports, Bluetooth, debug interfaces, as well as on products that are actively networked.
- Unfinished or non-machine-readable SBOMs are verified as triggers for RTA decisions and technical screenings under the 2025 eSTAR submission framework.
- Since the FDA enforcement trend of 2024-2025, submissions lacking the demographic performance breakdown (race, sex, age) are flagged as prejudiced, which is not only an ethical issue anymore, but a regulatory issue also.
The New Age of AI in Medicine
The 2026 Landscape: From Static Tools to Adaptive Partners
We formally exited the experimental phase of medical artificial intelligence early in 2026. Artificial intelligence and machine learning (AI/ML) are now integral components of hospitals. In modern medicine, they are the main part of the nervous system. Medical devices have changed from fixed decisions to data-driven clinical intelligence.
From an early oncology detection diagnostic tool to a triage system for cardiac emergencies, artificial intelligence-powered software as a medical device (SaMD) is now the norm in business. High-performance healthcare counts on these instruments to analyze data more quickly than any person could. With this change, computers become more active viewers rather than passive ones. It actively helps to save lives. Developers must now consider how their programs learn over time rather than just how they work on day one.
The 510(k) Dominance: Why 97% of AI Devices Choose “Substantial Equivalence.”
The figures indicate a clear story if you are building an artificial intelligence (AI) medical tool. According to contemporary FDA tracking, about 97% of AI-enabled medical devices approved for the U.S. market followed the 510(k) process. This supremacy results from the 510(k) pathway being far quicker than other possibilities. It enables developers to show that their application is largely equal to an existing market-prediction device.
Many times, this route lets you get past the multi-million dollar, multi-year clinical trials needed for Premarket Approval (PMA). For the rapid-paced software industry, the 510(k) is the only practical lane for quick innovation. It enables businesses to give patients their goods in months rather than years. The digital health revolution we see today runs on this regulatory plan.
This manual offers the definitive plan for negotiating the difficult 2026 legislative landscape. Your premarket filing of artificial intelligence software will be faultless and review-ready. You will know the steps necessary to convert your code into a cleared medical device by the end of this article.
Understanding FDA Rules for AI-Based Samd
The FDA does not work in isolation. It collaborates with the International Medical Device Regulators Forum (IMDRF). This partnership guarantees that several nations’ global SaMD policies are in line. For companies wanting to grow beyond the U.S. market later on, this is especially useful.
IMDRF’s Function and Risk Categorization
Based on two key features, the IMDRF framework classifies SaMD. First is the patient’s condition: vital, serious, or non-serious. Second comes the relevance of the data given by the program. Does it just educate or treat, diagnose, and drive clinical management? This matrix assists the FDA in determining the level of review your artificial intelligence calls for throughout the evaluation process.
Key FDA Guidance Documents Updated 2025–2026
You have to coordinate your development with three particular cornerstones to get an AI medical Device FDA approval.
- GMLP (Good Machine Learning Practices): The ethical guideline for artificial intelligence creation is GMLP, good machine learning practices. It encompasses anything from data quality to model tweaking.
- PCCP (Predetermined Change Control Plans): This is the plan for how your artificial intelligence will learn and evolve once it debuts. PCCP (Predetermined Change Control Plans). In 2026, the FDA is paying great attention to it.
- QMSR (Quality Management System Regulation): This is the updated 2026 standard that at long last fits FDA expectations with the international ISO 13485 norm.
- AI-Enabled Device Software Functions – Lifecycle Management and Marketing Submission Recommendations (Draft Guidance, January 6, 2025): The FDA has offered its most detailed guidance on AI SaMD, covering the complete lifecycle of the devices, including training data and pre-market validation, all the way to post-deployment oversight. It is necessary and mandatory to apply AI-enabled devices, and they should be applied together with all other relevant guidelines. In 2026, any submission that disregards this document will be taken to be outdated.
FDA’s Perspective on AI Lifecycle Management
The FDA no longer regards artificial intelligence as a one-and-done approval. They view it as a complete product lifecycle. This calls for preparations for how the model will work two or three years from now. They see how you manage fresh data and your monitoring programs. Compliance in 2026 is about ongoing quality, not just a one-time shot.
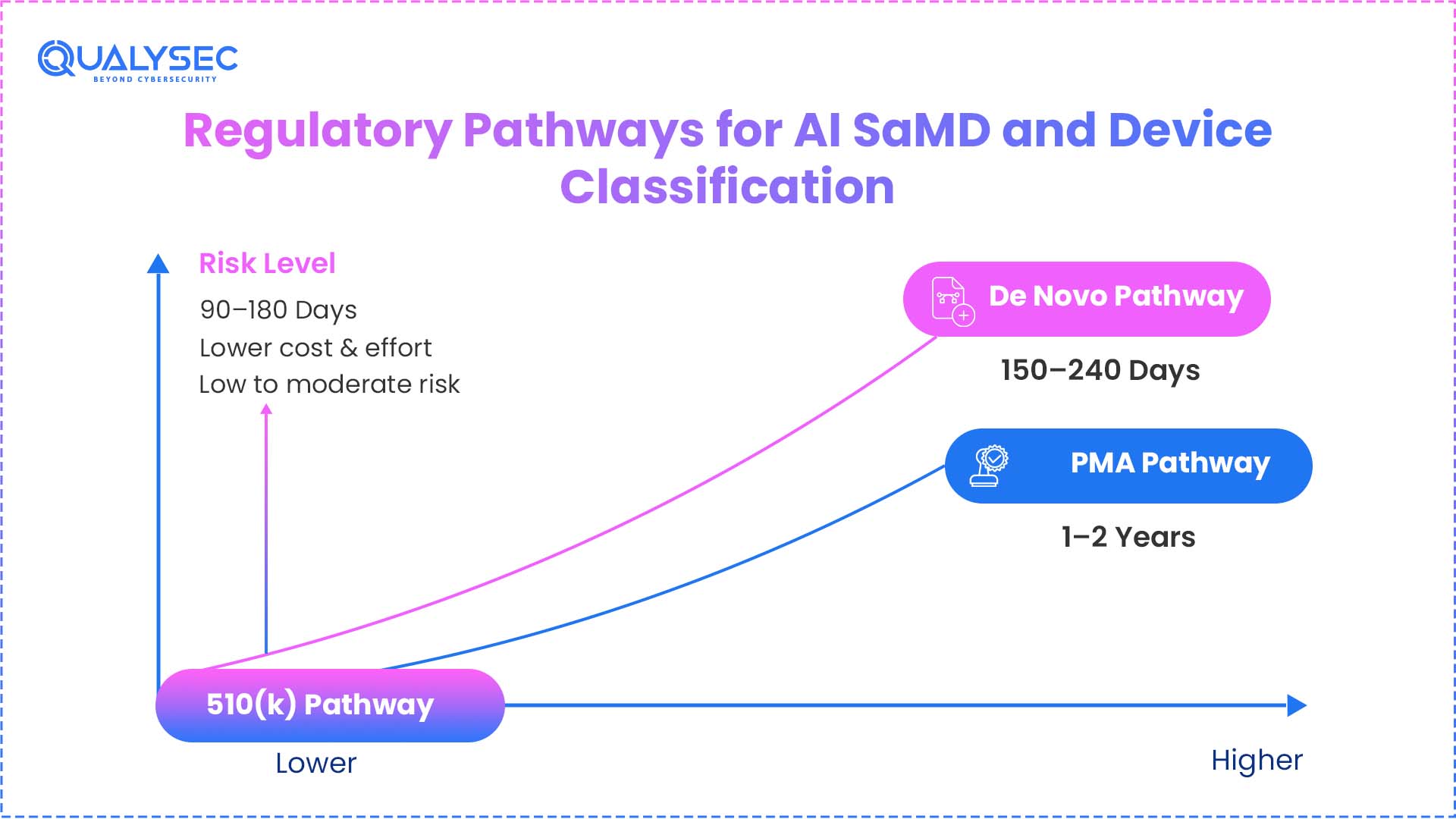
Regulatory Pathways for AI SaMD and Device Classification
The heart of a 510(k) is knowledge of significant equivalence. You are not showing that your artificial intelligence is brand new. Rather, you are demonstrating that it is as safe and efficient as something currently approved by the FDA.
- Artificial intelligence (AI) classification and FDA risk categories: Because of its complexity, artificial intelligence frequently elevates a device into a more dangerous classification. Adding artificial intelligence can elevate a manual tool from Class I to Class II. You must thoroughly assess how your artificial intelligence elements alter the risk profile of the entire system. Your risk assessment comes under FDA review first during the submission.
- Selecting the Correct Predicate: Legally marketed equipment against which you compare your product is called a predicate. Your predicate need not be artificial intelligence, really. One can match a manual, non-AI imaging workstation to an AI-based lung nodule detector. It works as long as the clinical goal is the same. Choosing the best predicate is your most critical strategic decision.
- When 510(k) Is Appropriate versus De Novo or PMA: You would have to travel the De Novo path if there is no predicate for your next innovation. This fundamentally generates a new categorization for your equipment. Though it takes longer, it enables you to be first in a new sector. PMA is set aside for items that have no equivalent equipment or are dangerous.
Feature | 510(k) Pathway | De Novo Pathway | PMA Pathway |
Comparison | Substantial Equivalence | No Predicate Exists | Life-Sustaining/High Risk |
Review Time | 90–150 Days (Avg) | 150–300 Days | 1–2+ Years |
Clinical Data | Sometimes Required | Mandatory (95%+) | Always (Extensive) |
Device Class | Mostly Class II | Class I or II (Novel) | Class III (Life-Sustaining) |
How AI and Machine Learning Are Transforming Medical Devices
Conventional medical software implements code written by the programmer. The 2005 ECG algorithm takes an interval that is measured and tries to match it to a hardcoded threshold, and the flag is returned. It is like that every time. The regulative power and the clinical ceiling of it is that predictability.
- The trade-off in deep learning is different. A model trained on hundreds of thousands of patient histories can find patterns that no threshold-setting programmer expected to find, but its explanations are not a readable rule set. The fact that such opaqueness is precisely the reason why the FDA formulated its GMLP principles and the January 2025 draft guidance on AI lifecycle management. The rules grew due to the technology compelling them to change.
- This has a direct effect on developers, its practical ramification being as follows: FDA does not assess AI SaMD at the time of submission anymore. It assesses the long-term safety and accuracy of the device, as well as in the population of patients that might not be similar to the training set, on hardware that the developer has no control over. An algorithm that is locked (that is, the weights of the algorithm are set in stone when it is deployed) is simpler to check but progressively harder to retain clinically useful. An adaptive algorithm is able to keep up with, but must be updated safely by a PCCP. It is that tension that the majority of the 510 (k) process on AI is, in actuality, dealing with.
Regulatory Frameworks for Evaluating AI Submissions
Several methodologies guide the FDA’s evaluation of your artificial intelligence. Unwatched AI models know they can change or degenerate over time. This resulted in the Total Product Lifecycle (TPLC).

Step I: Planning Your Predicate Strategy (Identify and Plan)
Your first major challenge is the substantial equivalence enigma. You have to demonstrate that any technological variances do not bring up new safety issues.
- Demonstrating Safety Against Non-AI Predicates: Should your predicate be a manually operated instrument, you have to prove that the artificial intelligence misses nothing a human would notice. You directly match your AI’s performance against the gold standard that doctors use now. Should artificial intelligence be just as good or better than humans? If so, then equivalence has been demonstrated.
- The Pre-Submission (Q-Sub) Meeting: You should never submit a 510(k) in 2026 without first receiving a Q-Sub. Before beginning formal testing, this is a gathering when you interact with the FDA reviewers. You might inquire of them whether your predicate is suitable and if your testing approach is adequate. This prevents you from receiving a rejection letter six months down the line.
- Optimal Standards for Predicate Selection: Find a predicate as near to your intended use as possible. Choose a predicate for your artificial intelligence that is for heart scans; that is, avoid picking a lung scan instrument. Look for recently authorized gadgets, as they follow the more contemporary, more pertinent FDA regulations.
Step II: Use Good Machine Learning Practices (GMLP)
Ten guiding principles for GMLP are stressed by the FDA. Following these is not optional if you want a smooth approval process.
- Curation and Data Quality: You have to show that your training data reflects the patients that the artificial intelligence will really encounter. A 2025 study, for instance, found that AI taught exclusively to one ethnic group failed more often when deployed in different hospitals. The FDA will examine your data to see if it encompasses a variety of races, sexes, and ages.
- Independent Validation and Overfitting: You have to keep your test set absolutely apart from your training set. Your results are fraudulent if your artificial intelligence examined the test data during its training stage. This is overfitting. To guarantee the results are truthful, the FDA calls for a locked vault approach to your final testing results.
- Algorithm Transparency (The Black Box): You have to show how the algorithm deals with data. You must offer some degree of explainability, even if it’s a complicated neuronal network. Inform the FDA of the characteristics the artificial intelligence is examining. They don’t like black boxes where nobody knows how the solution was found.
- Human-in-the-loop (HITL): Your artificial intelligence should assist the doctor, not substitute them. Create your system such that a human still makes the ultimate decision. A device including a human-in-the-loop safety check is substantially more likely to be approved by the FDA. This lowers the likelihood that the artificial intelligence will alone make a disastrous mistake.
Step III: Building the Foundation—The QMSR & ISO 13485:2016
The Food and Drug Administration’s (FDA) updated Quality Management System Regulation (QMSR) officially takes effect on February 2, 2026. For software developers, this is a significant revolution.
- Putting the 2026 QMSR into action: The ancient QSR guidelines have disappeared. Today, the FDA adheres to the international ISO 13485:2016 standard. This implies that your quality system has to be geared much more toward risk management. This bodes well if you already conduct sales in Europe. Otherwise, you have some documents to update.
- Control Designs for Artificial Intelligence: Data management is the new hardware engineering in the artificial intelligence industry. You have to approach your training datasets like real-world components. They need quality inspections, version numbers, and storage regulations. The FDA expects your design controls to span the whole data pipeline, not just the final code.
- Approaches Based on Risk: Risk-based decisions are needed throughout the whole organization under the updated QMSR. This is not only for the engineers. It’s for the clinical testing practitioners and those working with data. To prevent an audit failure, you must coordinate your software development life cycle with these new worldwide standards.
Qualysec provides the mandatory compliance auditing and gap analysis required to align your legacy QMS with the 2026 FDA standards.
Step IV: Build the 510(k) Submission Dossier
Pre-market submission of your artificial intelligence program is an avalanche of facts. It must be arranged exactly so the reviewer can find what they want.
- Comprehensive Device Description: This is not just a summary. This is a thorough examination of your artificial intelligence’s structure. You have to describe the architectural structure, the inputs, and the outputs. You must also include every clinical claim you intend to make in your marketing materials.
- Performance Testing and Validating: Real-world data is required to show performance. Prospective, multi-site investigations are highly regarded by the FDA. This implies you examined the artificial intelligence across several hospitals under many doctors. This shows the program operates in the untidy actual world rather than only on your PC.
- Cybersecurity and the SBOM: By 2026, you have to present a Software Bill of Materials (SBOM). Here are all the third-party libraries or open-source components in your code. The FDA wants to see your plan for rapidly fixing a vulnerability discovered in an open-source tool you used.
- Labeling and Instructions for Use (IFU): Your label must plainly specify the limitations of the gadget. Who is it for? For whom is it not? What well-known blunders could it possibly produce? The FDA wants you to be honest with the doctors who will be using your instrument.
Step V: Mastering the PCCP—Managing Algorithm Change
The locked algorithm is dying gradually. You have to include a predetermined change control plan if your artificial intelligence is to remain pertinent in 2026.
- The Death of the Locked Algorithm: Should your artificial intelligence not pick up, it will lag. A PCCP lets you update your model without having to file a new 510(k) each time. It is essentially a pre-approval for upcoming changes. Keeping an AI model correct depends on this technique as medical understanding evolves.
- Protocol Design: You have to be specific about how you will retrain the model in your PCCP. What information will you draw on? What counts as success? The new model will not be launched if it does not reach those figures. The FDA supports the method of modification, not only the modification itself.
- The Concept for Playgrounds: Consider the PCCP as demarcating a playground. Play (update) all you want as long as your artificial intelligence stays inside the fence—that is, the accepted procedure. You must return to the FDA for a fresh examination if it goes beyond the barrier. This enables quick updates while keeping the software safe.
Step VI: Valid Evidence and Clinical Data Package
Three main measures are of interest to the FDA. Your artificial intelligence medical device must reach these figures to be approved by the FDA.
- Sensitiveness, Specificity, and AUC: Sensitivity is about locating the illness. Specificity is about not crying wolf. Your artificial intelligence’s overall report is the AUC, or Area Under the Curve. You have to showcase studies demonstrating that these numbers are great enough for clinical applicability.
- Performance of Human-AI Team: The FDA is not only concerned about the code. They are concerned about how the code is used during a doctor’s performance. The performance of the physician may actually decrease if the AI is 99% accurate, but it mistakes the doctor. You have to demonstrate that artificial intelligence actually aids humans in making better decisions.
- Actual-World Evidence (RWE): You should continue accumulating data after your release. Your next 510(k) can be supported using this real-world evidence, or your PCCP’s performance can be proven. The FDA is starting to consider data from actual hospital use to demonstrate that a device is safe and effective.
Step VII: The “Black Box” Problem—Transparency
For 2026, explainability is the huge buzzword. Doctors won’t utilize artificial intelligence unless they grasp why it’s instructing them to do something.
- Logic in Communication Algorithm: Design human-readable artificial intelligence. Normally, this entails including a dashboard that displays which blood markers the AI employed to make its decision or which components of a scan. The AI should circle the suspicious zone so the physician can see it as well if it raises a tumor alert.
- Marks for Faith: The training data must be very clear on your label. Should your artificial intelligence be trained only on elderly people, the label must state so. Transparency helps doctors develop faith. Furthermore, it safeguards you against legal issues. Should artificial intelligence be applied to a patient kind for which it was not intended?
- Mitigating Bias: You must present evidence proving your artificial intelligence is just. The FDA is extremely rigorous regarding gender and racial prejudice in 2025 and 2026. Your artificial intelligence has to be shown to be as effective for a 30-year-old man as it is for a 70-year-old lady. Should there be a performance gap, you have to explain the causes as well as how you are addressing them.
Step VIII: Usability and Workflow Integration
In many cases, the failure of artificial intelligence is more of a usability problem and not a code problem. It will not be utilized in a proper manner in the event that the program is too complex to work with.
- Automation Bias: It is when a physician begins to have excessive confidence in artificial intelligence. All they do is approve and halt at the end of each two clicks of verification of results. Your interface needs to be created to counter this. There are times when it is a physical confirmation of the most significant conclusions of the artificial intelligence by the doctor.
- Human Factor testing: You must carry out research in a real-life setting. The FDA would be interested to learn how artificial intelligence works in a busy and noisy emergency room. The fact that the buttons are too small or the alerts too loud makes it unsafe. The fact that your AI software was submitted prior to the market undergoing substantial usability testing.
Risk Management—The Cybersecurity & Toxicity Shield
In 2026, the target is the victim of the first order in terms of cyberattack in SaMD. The SaMD cybersecurity requirements have substituted the make or buy component of any 510(k) filing, as the majority of AI applications are connected to being cloud-based. The Consolidated Appropriations Act of 2023, i.e., in Section 524B, within the context of which the FDA is already acting, requires manufacturers to give a reasonable assurance that the equipment is not harmful. It is more than a checklist, but it is a living shield that you should construct around your algorithm.
SaMD Penetration Testing: Strategic Ethical Hacking
Penetration Testing for SaMD must be documented for eSTAR compliance. This requires the services of professional ethical hackers who endeavor to identify Model Poisoning risks or corrupt the training data. The FDA now mandates a report that includes the Exploit Path and reproduction methods of every vulnerability discovered. They want to witness that you have documented the remediation of high-risk problems, rather than simply identifying them. All connections should be tested under stress against contemporary risks like BOLA (Broken Object Level Authorization) or data leakage in the event of your artificial intelligence conversing with a mobile application or a cloud API.
Qualysec’s specialized medical device penetration testing identifies model poisoning and maps the exploit paths required by FDA reviewers.


































































































































































































































































































































































































































































































































































































































































































0 Comments